

Introduction
Since the advent of effective antiretroviral therapy (ART) in the 1990s, HIV has transitioned into a manageable chronic infection. Despite successful control of viral replication in plasma and a significant reduction in mortality, a growing body of evidence indicates that even “optimally treated” HIV-infected patients exhibit a high prevalence of non-AIDS defining illnesses, including cardiovascular, respiratory, neurologic, metabolic, renal, and liver disease, as well as various solid and hematologic malignancies.1–12 While the underlying mechanisms are complex, many of these chronic conditions are linked to a generalized state of chronic inflammation and persistent immune activation. It is well established that chronic inflammation can damage multiple organ systems over time, accelerating the aging process.9 These observations have led to the concept of “HIV and aging” or “inflammaging,” where virologically suppressed HIV patients on ART experience progressive and disseminated organ damage compared to matched HIV-negative individuals, with a faster progression to age-related organ dysfunction.2,4,5 A critical goal in the fight against HIV is to reduce chronic inflammation and organ damage, ultimately aiming to prolong survival, decelerate the aging process, and improve quality of life.
Interestingly, “Elite Controllers” (ECs) who achieve virologic and immunological control without ART still experience chronic inflammation and aging, suggesting that “inflammaging” may be at least partially independent from virologic factors and a direct consequence of the inflammation itself.13 From an evolutionary perspective, Lentiviruses may need to avoid accelerating the aging process in their host to persist longer, as aging decreases host survival. Therefore, HIV and SIV infection in non-natural hosts may be “failing” from an evolutionary perspective (Figure 1). Due to the complexities described above, multiple clinical trials, proof-of-concept studies, and theoretical research models have been published aiming to reduce inflammation and aging in HIV patients on ART. The results of these efforts have been reviewed extensively elsewhere14–42 and have been largely disappointing. Our own HIV group has contributed to the field by proposing the use of tetracyclines due to their anti-inflammatory properties,43 being a group of molecules also proposed in the control of HIV-associated neuroinflammation. The benefits of these clinical trials have been partial and not definitive, and some of the proposed theoretical models have yet to be tested experimentally. This area remains a focus of intensive research and interest in the academic environment.
The search for non-pharmacological (preferably nutritional) anti-inflammatory therapies has been a long-standing goal for researchers worldwide. Recently, there has been a surge in public and scientific interest in the potential health benefits of Intermittent Fasting (InF) beyond weight loss.44–49 From an evolutionary standpoint, mammals have developed specific mechanisms over hundreds of thousands of years to overcome periods of food and energy scarcity in the environment. These mechanisms trigger stress signals as an adaptive strategy for survival. Interestingly, similar mechanisms have been observed in nematodes and yeasts,48 indicating the ancient nature and conservation of these pathways across different species. These alternative metabolic pathways are extremely effective in overcoming cellular stress and starvation, maintaining high levels of mental acuity and physical endurance, and offering numerous other health benefits, including reducing inflammation, promoting anti-aging effects, and accelerating autophagy.50 Multiple animal studies of InF have consistently demonstrated increased lifespan, decreased inflammation, improved treatment of diabetes and other metabolic diseases like obesity, enhanced cardiovascular health, reversed fatty liver, and promoted numerous neurocognitive benefits (including neuroprotection against stroke) - these findings have been reviewed in detail in previous works by Mattson and Longo.48,50 Importantly, most of these studies have been conducted on animal models, primarily mice.
The sophisticated underlying molecular mechanisms of InF, which we refer to as “The Survival Mode”, were likely originally designed to function for only short periods of time while stressful stimuli were present or during periods of food shortage. However, theoretically, these mechanisms can be utilized intermittently to take advantage of their multiple benefits, restoring cellular homeostasis, decreasing inflammation, and potentially slowing the aging process. The application of this approach in the treatment of chronic inflammatory conditions appears promising and potentially limitless.

The primary objective of this review is to explore the possible health benefits of InF in well-controlled HIV patients on ART, with the ultimate goal of reducing chronic inflammation, the accelerated aging process, and the development of chronic age-related diseases. Another objective is to educate the general public and general practitioners about a new concept in the ART era, viewing HIV disease as a “Chronic Inflammatory Disease” similar to many others, placing less emphasis on virologic factors as determinants of morbidity and mortality.
This review will be organized based on the most significant pathophysiological events that we believe may be interrupted by InF. It is not the goal of this review to provide a detailed review of the possible mechanisms of chronic inflammation in HIV; this has been reviewed in detail previously by our group.51
Important Note: This is a theoretical model that has not yet been proven and is based purely on theoretical speculation after an extensive literature review and cross-referencing metabolic and molecular pathways between HIV/SIV and intermittent fasting. This model will need to be tested in clinical trials.
1. Introduction to HIV-Associated Gut Mucosal Dysfunction: The Goals of Reversing Dysbiosis and Promoting Epithelial Gut Integrity
For a more detailed description of the role of the disruption of the gastrointestinal mucosa on HIV pathogenesis see our recent review: (https://nortonhealthcaremedicaljournal.scholasticahq.com/article/84063-hiv-and-aging-hiv-seen-as-a-chronic-inflammatory-intestinal-disease).
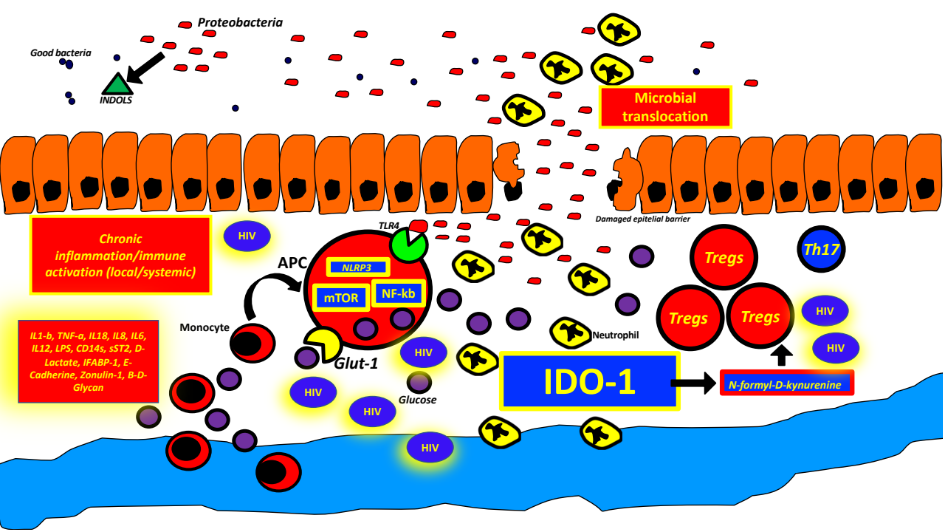
There is overwhelming evidence that persistent microbial translocation from a dysfunctional and structurally abnormal gastrointestinal mucosal barrier plays a primary role in the pathogenesis of HIV/SIV infection in non-natural hosts.52–60 Epithelial damage and microbial translocation are of paramount importance as the initial triggers of chronic inflammation and have been reviewed in detail elsewhere.61–66 Some of these mucosal changes occur early in the course of infection and are only partially reversed by ART. It is known that HIV specifically targets CCR5+CD4+ cells in the GALT.67 Importantly, intestinal lymphocytes represent over 60% of the total pool in both humans and non-human primates (NHP).68 There is a massive loss of CD4+ cells in SIV-infected RMs (Rhesus macaques) in the first 7 days post-infection, with 90% being wiped out within 2-3 weeks (mainly activated memory cells).68 These early mucosal changes are so crucial in the pathogenic HIV/SIV infection model that we believe we should consider chronic SIV/HIV infection in non-natural hosts, at least in part, as a “Chronic Gastrointestinal Inflammatory Disease”. In fact, there are multiple similarities between Inflammatory Bowel Diseases (IBD) and HIV regarding microbial translocation and chronic systemic inflammation.61
Importantly, during IBD, there is no depletion of mucosal CD4+ cells; on the contrary, there is an increased influx of these cells, including Th17 cells,68 suggesting that gut inflammation and microbial translocation alone, without viral replication, are not sufficient to cause mucosal CD4+ depletion.
In a previous review,51 we proposed that, through years of evolution, SIV or HIV in non-natural hosts have learned how to artificially create “gaps” in the tight junctions between epithelial cells, ultimately aiming to feed off local inflammation and promote local viral persistence, a key objective of the Lentivirus family.51,69 A recent experiment with Caco cells demonstrated that HIV-1 itself, through its gp140 protein, can cause these changes early during infection, reducing Claudins (CLDN), Occludins (OCLN), and Zonulin (ZO-1) - these changes can be reversed with the administration of IL17A-F.69 In another study, plasma levels of IFABP and sST-2 were elevated in a cohort of 48 recently infected HIV-positive patients, confirming the early onset of enterocyte damage and release of structural proteins.63 There is a decreased amount of de novo synthesis of ZO-1, Claudin 1-2, and E-cadherin early after the initial infection. To support this, a revealing recent study using proteomic analysis showed that as early as 3 days after SIV infection there was a decrease in proteins related to epithelial integrity, and that 14 days after infection, there were increases in proteins related to the innate immune system, providing evidence that epithelial damage occurs before the innate immune response.64 In NHP models of SIV infection, as early as 14 days after primoinfection, there is an almost complete depletion of CCR5+CD4+ T memory cells, which persists if left untreated.68
In fact, an increase in plasma REG3-alpha (a marker of gut damage) is elevated in PLWH (whether on treatment or not), including elite controllers, compared to HIV-negative controls. REG-3-alpha levels correlate inversely with CD4 cell count and positively with HIV viral load.70
On the other hand, natural hosts of SIV (Sooty mangabeys, African green monkeys, Madrills, Drills, and Suntailed monkeys) do not exhibit chronic inflammation, microbial translocation, immune activation, immune exhaustion, AIDS, or accelerated aging.71 This indicates that HIV/SIV can persist in non-natural hosts but at the expense of creating inflammation and tissue damage, ultimately accelerating aging and decreasing the lifespan of the host. The CD4+ cells of natural hosts experience an initial slight decrease after the acute infection, but soon after, pre-infection levels are reached with a downregulation of CCR5, which reduces de novo infections.68 Natural hosts have adapted and learned how to control cellular depletion and inflammation, one known mechanism being the decrease in the number of HIV-target cells, like CCR5-CD4 T lymphocytes, in the GALT.68
Intriguingly, if you give high-fat diets to natural hosts of SIV (African green monkeys), they develop progressive disease with increased microbial translocation, immune activation, viral replication, and expansion of reservoirs72 which may imply that microbial translocation / inflammation may trigger accelerated viral replication.
SIV in natural hosts significantly decreases inflammation and immune activation after the transition from acute to chronic SIV infection, despite persistent robust viral replication, highlighting the fact that viral factors may not be as important as immunomodulatory factors for CD4+ depletion and disease progression.68 This has been achieved through enhanced gut mucosal repair mechanisms associated with decreased microbial translocation. Also in natural hosts, a mutated gene of ICAM2 and TLR-4 impairs the response to LPS stimulation, subsequently decreasing the production of TNF and IL6 and local/systemic inflammation.73 Also, sooty mangabeys’ macrophages (SIV-natural host) were found to be more resistant to infection by SIV than their rhesus macaques’ counterparts (SIV non-natural hosts) through increased expression of tetherin and TRIM22.74 Another novel mechanism of protection against SIV in natural hosts is the downregulation of interferon responses, which is associated with pathogenic SIV and HIV, creating immune activation, immune exhaustion, and apoptosis.68
ECs (still, non-natural hosts), even though they have immunological and virologic control without ART, still have some degree of epithelial damage since their IFABP-1 levels in plasma are lower compared to HIV progressors, but still higher than HIV uninfected controls.75 This implies that viral factors alone may not be sufficient to explain the persistent inflammatory state in elite controllers, reinforcing the concept of “Collateral Damage” in order to persist. From this perspective, chronic inflammation is a non-desirable consequence to persist in an inflammatory environment.
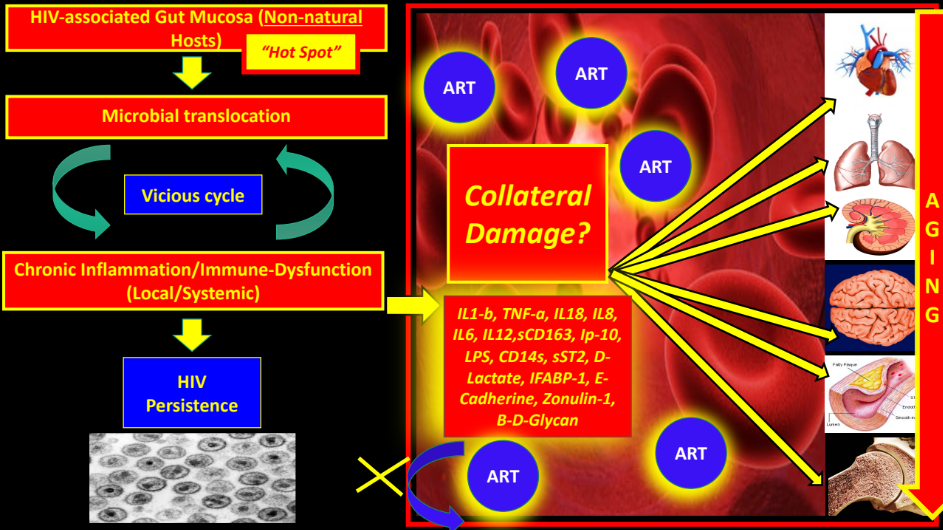
In summary, this artificially created “Hot Spot” (as we call the GUT-mucosa and GALT - HIV/SIV-associated changes) soon after infection is continuously fed with an enormous amount of antigens, chemicals, bacteria, fungi, viruses, and environmental substances, which, in turn, promotes persistent activation of the innate immune system in the context of an already abnormal microbiome (see below “Dysbiosis”). Bacterial and fungal (and possibly viral) antigens continuously cross-over through the gaps without immunological control, even after being on ART. In fact, it is possible to measure increased D-Lactate, IFABP-1 (Intestinal Fatty Acid Binding Protein), REG3-alpha76 (also increased in IBD), and sST2 in HIV patients well-controlled on ART. Of note, IFABP-1 has been linked to increased mortality and CD4+ decline in HIV.75 Interestingly, a recent study found that circulating Beta-d-Glucan (BDG) has a better correlation with cardiovascular disease (measured with cardiac computed tomography) than LPS.77
A rapid decline in Th17 cells (CCR6 positive) after acute SIV/HIV infection with an increase in the ratio of Tregs/Th17 is believed to be the culprit for the persistent lack of epithelial homeostasis and lack of repair of the tight junctions after the initial damage.61,63,65,69,71,78,79 Importantly, Tregs are also important reservoirs for HIV.80 An increased Th17/Tregs ratio is seen in pathogenic infections with HIV/SIV and is linked to inflammation and immune activation.68
The Th17/Treg ratio remains stable during chronic SIV infections in natural hosts, while it is decreased in pathogenic HIV/SIV infection in non-natural hosts and predicts disease progression.68
T.regs increase “tolerance” to microbial product leakage, as their main function is to suppress (“regulate”) the immune response, usually against self-antigens.63 The mechanism used by HIV-infected T.regs to regulate immune activation may be mediated through the generation of local adenosine (“Purinergic pathway”).80 Unfortunately, Th17 cells are only partially restored with ART in the blood and not restored at all in the GALT (Gut Associated Lymphoid Tissue), suggesting an irreversible phenomenon.65 As a result, pathogenic HIV/SIV can decrease the production of IL-17 and IL-22, which are highly involved in the repair of the tight junctions and barrier integrity.68 IL-21-producing CD4+ T lymphocytes are also depleted during SIV infection, which can alter immune pathways favoring Th17 differentiation.68 Activation-induced pyroptosis is responsible for the loss of MAIT cells in the GI mucosa of PLWH (even on ART) compared to HIV-negative controls.81 MAIT cells are responsible for the integrity of the epithelial barrier against external challenges, such as microbial or fungal translocation. In the same study, the absolute number of MAIT cells correlated inversely with plasma sCD14+ (marker of monocyte activation due to microbial translocation) and I-FABP levels in HIV treatment-naïve patients.
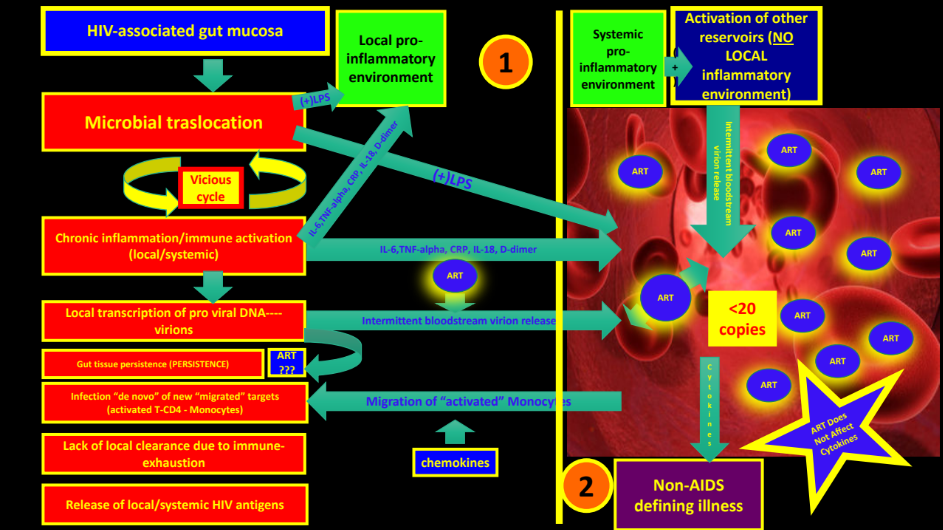
The above phenomena represents a “vicious cycle” in which persistent microbial translocation of an already dysbiotic microbiome through a “leaky epithelium” continuously activates an already exhausted and senescent innate and adaptive immune system (Figure 2 and 3).
.png)

The above mechanisms generates chronic inflammation and immune activation with local HIV persistence as immune cells are constantly activated, generating new HIV targets. Probably, ART, even though it may not penetrate the GUT mucosa and GALT very well, is effective enough to control gross active HIV replication in plasma. Importantly, inflammatory signals (interleukins, chemokines, etc.) and microbial products have already reached the bloodstream at this point and will deliver the initial inflammatory trigger (“spark”) in distant organs (CNS, Cardiovascular, Renal, Bone, etc.), accelerating the aging process and organ damage. Maintenance of epithelial gut integrity with enhanced repair mechanisms in SIV-infected RMs with subsequent lack of microbial translocation and immune activation may explain how a functional cure may be reached in pathogenic SIV/HIV models.
2. Intersection Between HIV/SIV Pathways and InF
2.1. The “Aging” Process, Telomere Length, the Mammalian Target of Rapamycin (mTOR), and Autophagy
Immune cells in well-controlled HIV patients exhibit characteristic phenotypic changes compatible with “immune senescence” or “immune exhaustion”, rendering them dysfunctional.2,4,5 Cellular senescence is a hallmark of the aging process. It is well-established that even well-controlled HIV patients on ART age at an accelerated rate (see above). When we mention “accelerated aging,” we refer to a higher prevalence of non-AIDS defining illnesses (cardiovascular, respiratory, neurologic, metabolic, renal, and liver disease) compared to age-matched HIV-negative control subjects.1–12 There is a clear link between inflammation and aging. One theory is that chronic inflammation generates reactive oxygen species (ROS) that damage telomeres, increasing cellular senescence and aging of different organ systems. Telomere Length (TL) has traditionally been linked to senescence and aging; shorter telomeres indicate a more accelerated aging process. A recent revealing study showed that even HIV patients on ART have shorter TLs, and a significant association between TL and inflammatory markers, like CXCL1, TGF-α, and IL10RA, was documented.6 PLWH have shorter telomeres on HSCs (Hematopoietic Stem cells), monocytes (CD14+), and leukocytes compared with age-matched controls.13 An inverse relationship was observed between myeloid cell activation (sCD163) and telomere length, suggesting that inflammation may shorten telomere length.13 This means that chronic inflammation and immune activation can shorten TLs, increasing aging most likely through ROS generation and DNA or protein damage in targeted tissues.
In addition to the possible effect of InF on TLs, a study in mice recently showed that InF can affect DNA methylation, potentially opening a window to its anti-aging properties.82 Studies have shown an increase of age-associated DNA methylation in PLWH,13 which is partially restored with ART. HIV accelerates aging (DNA methylation) and ART only partially slows down the accelerated process.83
HIV patients typically show an increased proportion of terminally differentiated cells in the CD4 and CD8 pool with markers of immunosenescence like CD57(+).12 Other markers of senescence during HIV infection include decreased numbers of naïve T cells [which can be partially due to thymic dysfunction12], increased TNF-alpha, sCD163, and sCD14.13 The increased proportion of terminally differentiated cells is somewhat disadvantageous, as it may indicate a decrease in the number or functionality of the pool of stem cells. In fact, stem cells also show decreased TL as they age. One study showed shorter telomeres in young HIV+ males compared to aged controls,13 regardless of whether or not they had virologic control with ART.
As a general concept, stem cells should have longer telomeres compared to differentiated cells. In fact, TL has been traditionally used to localize stem cell compartments in mouse studies.84 Pluripotential Stem cells (PSCs) are important players in the armamentarium against aging, as seen in Planarian organisms (a planarian is a flatworm of the class Turbellaria known for its permanent capacity to heal).84 Interestingly, a relationship was documented in this organism between mTOR, telomere length, calorie restriction, and stem cell repopulation-enrichment. The pool of mostly enriched stem cells reached the longest TLs through inhibition of mTOR during fasting.84 Therefore, it appears that, at least in planarians, starvation enriched the telomere length in PSCs through mTOR inhibition, offering a potential anti-aging strategy to explore in HIV patients.
The Mammalian Target of Rapamycin (mTOR) integrates input from upstream pathways, sensing cell nutrition, oxygen, stress, and energy levels. It regulates metabolism and physiology and is regulated by growth factors, hormones, cytokines, and stress. Importantly, mTOR is linked to accelerated aging, mainly through inhibition of autophagy. mTOR could easily be the link between chronic inflammatory diseases and accelerated aging. To understand the crucial role that mTOR plays in the accelerating aging process of HIV patients on ART, some concepts need to be clarified:
In HIV/SIV pathogenic models, leaked microbial products, mainly LPS (Lipopolysaccharides), strongly stimulate the innate immune system locally as follows:
-
LPS binds the LBP (Lipid Binding Protein), which stimulates TLR4 (Toll-Like Receptor-4) with the release of CD14s (measurable in plasma), a well-known marker of inflammation from microbial translocation.
-
TLR4 stimulates the mTOR through the MyD88 pathway in Antigen Presenting Cells (APCs), which, among its multiple functions, is the activation of the transcription factor NF-kb (see below under “NF-kb”).
Interestingly, HIV-specific CD8+ T cells from HIV-treated patients have higher expression of genes related to mTOR activation and glycolytic pathways compared with their counterparts from HIV elite controllers.85 It is intriguing that natural hosts for SIV do not experience immune activation and chronic inflammation or AIDS, even in the presence of high SIV viremia. Over thousands of years of evolution, a “blunted TLR4 response to ligands” through mutations in the TLR4 gene and decreased expression of ICAM-2 has been demonstrated in SIV natural hosts.73
-
Importantly, fungal products like Beta-D-Glycan (BDG) also leak through the dysfunctional epithelium, and it is well-known that activation through TLR 3 to 9 also stimulates mTOR through the MD2 – MyD88/PI3K/AKT pathway, with the same consequences as with bacterial products.
-
At the level of this dysfunctional mucosa (“Hot Spot”), the mTOR remains transcriptionally active due to constant microbial stimulation.
-
mTOR activity can also be increased in local CD4 T cells by IL15 and IL7 through PD-1 mediated pathways, which, in turn, will promote HIV immune escape and cellular HIV persistence.
-
Persistent activation of mTOR will promote further inflammation and local HIV persistence with an immune-metabolic switch of immune cells to a pro-inflammatory glycolytic phenotype (see below under “Warburg-like Phenotype”).
There are multiple studies demonstrating the importance of mTOR in HIV pathogenesis. It was shown that peripheral blood mononuclear cells (PBMC) of HIV-1 controllers present significantly higher amounts of autophagic vesicles compared to normal HIV progressors. Importantly, ex vivo treatment of those PBMC from HIV-1 controllers with Rapamycin (which inhibits mTOR1) results in an even more efficient autophagic response.86 These data support the hypothesis that autophagy may play a significant role in achieving virologic and immunological control without ART in HIV elite controllers. It has also been proven that targeting cellular mTOR with INK128 (mTOR inhibitor) decreased CCR5 expression and decreased the transcriptional activity of HIV,87 demonstrating the importance of silencing mTOR in any future attempt to achieve a functional cure along with ART in HIV patients. Of note, in HIV pathogenesis, mTOR will promote inflammation (through NF-kb activation) and aging (inflammaging), with its activation being detrimental for HIV control. The importance of autophagy for HIV control was shown in another study where Dactolisib and other mTOR inhibitors all decreased HIV replication in macrophages in a dose-dependent manner via degradation of intracellular HIV through autophagy without apoptosis. Interestingly, inhibitors of autophagosome-lysosome fusion and of lysosomal hydrolases all blocked the inhibition of HIV. This demonstrated that the inhibition of PI3K/MTOR and PI3K/MTOR/BRD4 for HIV control requires the formation of autophagosomes as well as their subsequent maturation into autolysosomes.88 These studies highlight the need to silence mTOR in order to potentially control HIV replication through autophagy. This opens a new door in the fight against HIV using ancient evolutionary mechanisms to clear not only damaged molecules but also cellular intruders like HIV.
2.2. Possible Mechanisms of InF to Overcome mTOR and Aging
Lessons learned from simple cell organisms and animal studies indicate that InF increases lifespan with overwhelming evidence.48 A 15-20% increase in longevity and a 65-90% reduction in tumor burden were achieved in Wistar rats after intermittent fasting strategies were applied in an experimental study.46 Interestingly, it appears that “Acetate”, which is elevated during fasting, is implicated in these anti-aging effects (see below under “SCFAs”). Rats lived twice as long when exposed to InF compared to rats fed ad libitum.50 This alternate metabolic pathway to optimize energy in the absence of food originated hundreds of thousands of years ago and has been highly conserved through different species. InF promotes a “Survival-mode” when nutrients are lacking in the environment or when signals of cellular stress are detected. This mechanism was probably not only created to use alternative sources of energy but also to clear cells of toxic molecules, ROS, DNA damage, cellular debris, misfolded proteins, and potentially intracellular organisms through autophagy. It is well-recognized that mTOR activation has “Pro-aging” effects (see above) and favors senescence later in life, as it inhibits autophagy. mTOR is thought to be responsible for the cellular accumulation of damaged organelles, oxidized proteins, and cellular debris, as it inhibits the clearance of these molecules. In general, the anti-aging mechanisms of InF are thought to be due to:
-
improved cellular stress adaptation (mainly protecting against ROS generation)
-
decreased inflammation and DNA damage
-
improve mitochondrial biogenesis
-
decreased mTOR expression (see above)48
-
promoting autophagy.
Therefore, the main direct mechanism of InF against accelerated aging in HIV would be through inhibition of mTOR,89 similar to the effects of Rapamycin in some animal models.90–92 The inhibition of mTOR may increase autophagy, decrease inflammation and pro-glycolytic immune pathways, and potentially, over time, have some influence over the HIV size reservoirs along with ART (hypothetical theory).
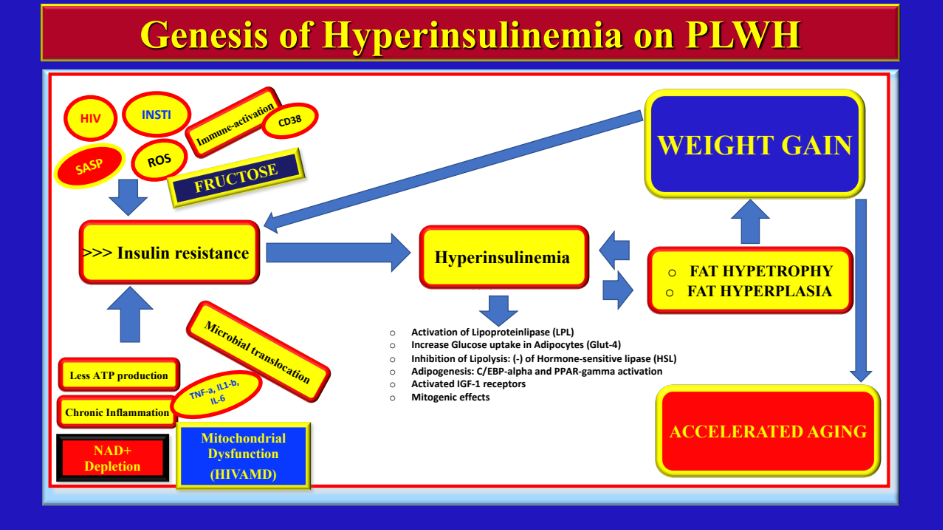
There may also be multiple indirect mechanisms with InF that may improve the local gut inflammatory environment (gut homeostasis) and decrease local and systemic inflammation. In fact, there is objective evidence of decreased amounts of oxidative damage to proteins, lipids, and DNA in the tissues of rodents exposed to InF,93 which may apply to human HIV subjects as well. Importantly, this could be significant, as there is ongoing protein and DNA damage (even mitochondrial DNA damage with certain NRTIs) in PLWH on ART with chronic inflammation that can potentially trigger cellular signals to activate the Inflammasome (see below under “Inflammation”) and generate further inflammation. Three days of fasting decreased the levels of Insulin and Insulin-growth factor 1 (IGF-1) in animal models, both of which are associated with accelerated aging and cancer formation.48
InF may also potentially improve or completely reverse the pro-inflammatory dysbiosis (with predominance of "Prevotella") seen in HIV patients on ART (see below under “Dysbiosis”). A reversal of the dysbiosis may decrease microbial translocation (bacterial and fungal) and the TLR4 overstimulation, which activates mTOR and promotes inflammation and aging. Also, the significant increase in "Short Chain Fatty Acids (SCFAs)" (see below under “SCFAs”) seen during InF may decrease local gut inflammation and may contribute to reversing the damage to the epithelial tight junctions. SCFAs may also have some impact on “purging” HIV reservoirs from latently infected cells, as they function as deacetylases inhibitors (see below under “SCFAs”). InF may also have some impact on the decreased generation of “Indols” (see below under “Indols”) produced mainly from Proteobacteria from tryptophan, which are pro-inflammatory. A favorable change or reversal of the dysbiosis may promote less Indole generation and less “IDO-1 activity” (see below under “IDO-1”).
InF may also inhibit the NLRP3 inflammasome (see below under “Inflammation”), and glycolytic pro-inflammatory pathways (see below under "Warburg-like phenotype). Overall, systemic inflammation should decrease through multiple mechanisms (directly and indirectly). InF may also regenerate the pool of Pluripotential Stem Cells through inhibition of mTOR as we discussed for planarians and, consequently, promote anti-aging effects, including regeneration of naïve lymphocytes (with potential immunological recovery), repair of epithelial cells, and decreased organ damage in general.84 The concept of enhancing PSCs with inhibition of mTOR during fasted states has been described previously.94 In fact, it has been shown that InF promotes hematopoietic stem cell activation and regeneration of immune cells.95
Interestingly, it was recently shown that during InF, Paneth cells in the crypts of the gut mucosa may promote the growth of Intestinal Stem Cells (ISCs) through inhibition of mTOR, a result that was also reproduced by providing Rapamycin (mTOR inhibitor).96
This could indicate that InF may have implications for epithelial and tight junction repair after initial HIV gut mucosal damage,94 as Paneth cells may sense the lack of nutrients as a signal of cellular stress. This study showed a link between nutritional status and stem cell function through mTOR inhibition, as mTOR controls ISCs function depending on nutritional factors.96 There is more evidence recently that ISCs favor the OXPHOS route instead of glycolysis for epithelial regeneration and that glycolysis actually disrupts their function.96 This could be important for gut epithelial regeneration in HIV patients during InF as OXPHOS will be favored, and therefore, we should expect an increase in the activity of ISCs. InF has also been shown to increase ISCs numbers, increase ISCs self-renewal, increase ISCs regeneration, and decrease ISCs differentiation.96
During InF, the FOXO transcription factor will be activated, which has been shown to protect stem cells, activating genes that are protective against oxidative stress and also promoting autophagy and stem cells self-renewal.96 FOXO activation promotes the expression of genes involved in antioxidant defense, DNA repair, and cellular stress responses. This helps cells withstand damaging conditions like oxidative stress, which is prevalent in HIV infection and general aging.
In summary, inhibition of mTOR, Insulin, and IGF-1 along with activation of the FOXO and OXPHOS pathways may promote the growth and self-renewal of ISCs in HIV patients, repairing epithelial damage (mainly tight junctions) and decreasing microbial translocation. It is clear that InF should have benefits in gut homeostasis, as it has been shown to limit intestinal toxicity during chemotherapy, probably reflecting increased activity of ISCs.96
3. “Warburg-like” Phenotype in Immune Cells: The Immune-metabolic Switch
Published evidence clearly indicates that the local gut inflammatory environment in HIV patients promotes glycolysis and the exocytosis of its products in immune cells.61–63,65,66,79,97,98 This phenomenon was initially observed in cancer cells, which, even in aerobic conditions, use glycolysis as the preferred source of energy, but it also occurs in immune cells when they are persistently activated. This phenomenon is called “Metabolic Reprogramming” and implies that immune cells undergo a “Broken Krebs cycle” with the accumulation of Citrate and Succinate, both of which are very pro-inflammatory. The final result is the activation of the NLRP3 inflammasome and its consequences99 (see below under “Inflammasome”).
Briefly summarized, the genesis of increased glycolytic pathways in HIV may be related to multiple factors:
-
An abnormal microbiome (“pro-glycolytic” dysbiosis)
-
Strict nutritional factors (diet rich in ultraprocessed food)
-
Preferred (selected through evolution) glycolytic pathways during inflammatory states
-
Combination of the above.
The process starts when, through molecular signaling, there is a constant recruitment of metabolically active monocytes (who express more Integrin B7) from the bloodstream to the “Hot Spot” (Gut-mucosa). The chemokine monocyte chemoattractant protein 1 (MCP1) or CCL2 is mainly responsible for this recruitment. These monocytes will overexpress Glut-1 transporters, which favors the accelerated internalization and overutilization of glucose. The immune-metabolic phenotype change also affects the lymphocyte subclass CD4+ PD-1 / CTLA-4, which, through mTOR pathways, will promote further local HIV persistence. The increased availability of glucose locally (intracellularly and extracellularly through exocytosis in vesicles) will also favor the phenotype switch of bystander immune cells (especially APCs and macrophages) to the M1 phenotype or “Warburg-like” (Glycolytic or Pro-inflammatory).61 The hyper-glycolytic environment will promote HIV persistence and a “hyper-pro-inflammatory local environment” with significant increases in the secretion of TNF-alpha, IL-6, and CXCL2.61 In fact, increased translocation of LPS and its uptake by these cells (mainly through TLR4 pathways) will promote the switch from Oxidative phosphorylation (OXPHOS) to glycolysis, providing proof of a metabolic-driven immune phenotype change.
This topic has been extensively explored in a recent review100 showing that the proportion of CD4+Glut1+ cells correlated with an increase in immune activation, as defined by the expression of HLA-DR and CD38, and inversely correlated with the absolute CD4+ cell counts.
There is a clear association between T cell glycolysis activity and increased susceptibility to HIV infection, which was demonstrated in multiple studies from different groups, and was proven by impaired viral infection due to the inhibition of glycolysis in some invitro studies. The potential therapeutic benefits of the “metabolic plasticity” (or "metabolic switch") offered by InF (mainly the switch from glycolytic pathways to lipid metabolism or OXPHOS) in the fight against HIV are undeniable.
InF will inevitably decrease the availability of glucose at the GI mucosa, with less interaction with the receptors Glut-1 in immune cells.
Intriguingly, immunometabolic changes were documented in HIV elite controllers with fewer metabolomes involved in glycolysis and the Krebs cycle and favoring lipid metabolism of specific HIV-1 CD8(+) T cells, making them more active and polyfunctional.85
Importantly, in the healthy gut mucosa of HIV-negative individuals, the predominance of Firmicutes (with a decrease in Proteobacteria) increases the local availability of Butyrate, which, in turn, due to its high volume of distribution, reaches immune cells and promotes lipid B-oxidation and an OXPHOS metabolic switch, with a subsequent increase in the release of anti-inflammatory and antimicrobial molecules (Oxidative-M2 Phenotype). HIV patients, however, present a local increase in Proteobacteria, Prevotella, and fungus (see below under “Dysbiosis”). Their products (LPS, B-d-Glucans) promote glycolysis, the secretion of pro-inflammatory cytokines, and further recruitment of Warburg-like monocytes from the bloodstream in a persistent, endless vicious cycle.61 At the same time, there is a correlation between glycolytic-inflammatory pathways and activation of the mTOR pathway, which will have therapeutic implications (see above under “mTOR” and “Aging”).
HIV itself, through its Vpr protein, can stimulate this immune-metabolic change through the HIF-1 alpha (stress response to hypoxia) and PPAR-gamma pathway.61 Some theories also point towards the potential for HIV itself to stimulate HIF-1 alpha pathways in lymphocytes with the secretion of glycolytic products promoting cell exhaustion to local bystander cells.61 It is important to note that a recent study linked a diet rich in saturated fats and carbohydrates with microbial translocation and increased IFABP-1 in plasma, suggesting that it may be possible to regulate the inflammatory response in the gut mucosa and control the metabolic switch of immune cells with nutritional strategies even in PLWH.75
In summary, glucose availability and glycolytic pathways are important players in the genesis and maintenance of a local and systemic inflammatory environment and HIV persistence.
3.1. Possible Mechanisms of InF to Overcome The Immune-metabolic Switch
InF could potentially prevent the above metabolic phenotype switch through multiple mechanisms:
-
Decrease Availability of Glucose: An interesting review about the potential application of fasting in cancer therapy indicated that InF prevents the conversion of Macrophages to the M1 phenotype and decreases the amounts of Tregs.101 This could be due to inhibition of the mTOR pathway in APCs, macrophages, or lymphocytes, or due to the lack of availability of glucose locally. This concept of " Starve the Cancer " could potentially be applied to HIV with prolonged and supervised fasting protocols. Currently, multiple trials are being conducted in cancer patients on chemotherapy, aiming to interrupt glycolytic pathways in cancer cells through InF. As metabolomic studies have shown that HIV progressors have a favored “glucolytic profile” of immune cells with impaired mitochondrial function, InF could counteract these mechanisms due to decreased availability of glucose (and glycolysis) and improved mitochondrial bioenergetics due to cellular adaptation to physiologic stress.48,50,93 If InF is effective, metabolomic analysis may show metabolic products predominantly originated from the OXPHOS pathways (mitochondrial).
-
Increased Butyric Acid and other SCFAs: InF will also promote a significant increase in the amount of local Butyric acid and other SCFAs, which may favor the switch to the M2 phenotype. Butyric acid will promote a local anti-inflammatory environment through the inhibition of certain cytokines (see below under “SCFA”) to overcome the hyper-inflammatory state of the phenotype switch (pro-glycolytic). Once the metabolic switch to a state of ketosis is reached, the local availability of Glucose will be significantly low, and SCFAs will be high, with a significant decrease in the exocytosis of vesicles packed with pro-inflammatory molecules (products of glycolysis) from immune cells.
-
mTOR Inhibition on Immune Cells: Even in the unlikely scenario that some immune cells still switch to the M1 phenotype (even with total low local availability of glucose), InF will still inhibit the mTOR pathway and promote autophagy of those cells. The inhibition of mTOR with InF will decrease the recruitment of Glut-1 transporters to the cell surface of immune cells, which would decrease glycolysis even further.
In summary, InF, through the decrease in the substrate (Glucose), decrease in the transporters (Glut-1), inhibition of the effector pathways (mTOR), and decreasing inflammation (SCFAs) will favor an anti-inflammatory environment with less generation of HIV target cells (activated immune cells) and less recruitment of activated monocytes.
- Microbiome Modulation: Finally, InF may promote a healthier gut microbiome with a possible phenotype switch to Firmicutes and Lactobacillus with detrimental effects on Proteobacterias and Prevotella. This reversal in the dysbiosis could, in turn, increase the amount of SCFAs (mainly Butyric acid and Propionic acid) and decrease the production of Indols (see below under “Indols”).
4. Inflammation and Cardiovascular Disease
Well-controlled HIV patients on ART still have significantly increased levels of inflammation and immune activation compared to HIV-uninfected controls, which is thought to be independent of viral replication (at least in plasma) and multifactorial.6,8,10,12,51 IL-6, C-reactive protein, TNF, IL-6, IFN-gamma, and D-dimer remain elevated after effective ART.65 It was even found that CD14’s (a surrogate for microbial translocation and monocyte activation) predicted mortality,65 highlighting the importance of decreasing inflammation as a therapeutic goal. Also, chemokines like IL-8, RANTES, CCL2, and Interferon gamma-induced protein 10 (IP10) remain elevated even after virologic control on ART.99 CXCL13 (a marker of immune activation) is elevated in PLWH compared to HIV uninfected controls and is correlated with HIV VL, CD4 T cell count, CD4/CD8 ratio, LPS, sCD14, (1→3)-β-D-Glucan, total IgG, TNF-α, Kynurenine/Tryptophan ratio (see below section IDO-1), and frequency of CD38+HLA-DR+ CD4 and CD8 T cells.102 In the same study, ART reduced CXCL13 levels without normalization. For more details about the possible sources of inflammation in HIV patients on ART, please review our last publication.51
Even ECs have increased systemic inflammation and immune activation thought to be caused by an increase in the frequency of γδ T cells in the gut mucosa in one study.103 Importantly, SIV natural hosts restore the CD4+ pool at the GALT mucosa after an initial massive loss during the acute infection through control of the inflammation and immune activation.68 Effective control of inflammation and immune activation at the GALT mucosal sites may have implications for reaching a " functional cure ".
To understand the pathophysiology of chronic inflammation in chronic pathogenic HIV/SIV models, some key players need to be defined:
-
The NLRP3 Inflammasome: This multiprotein platform is activated by infection or cellular stress. Its activation leads to caspase-1-dependent secretion of proinflammatory cytokines like interleukin-1β (IL-1β) and IL-18, and it leads to an inflammatory form of cell death termed “pyroptosis”.104 The inflammasome can be activated directly by HIV through TLR8 activation after contact with viral ARN,99 but also by other TLRs-mediated pathways (like TLR4 with LPS - see prior section). One theory is that the decrease in CD4+ cells could be due to apoptosis and pyroptosis, while immune activation is mainly mediated by pyroptosis.67
-
NF-kb Activation: This is essential for the NLRP3 / ASC / Pro-Caspase complex activation. NF-kb can also be activated by stress, TNF-alpha, IL1-b, free radicals, HIV itself, LPS, oxidized LDL, and bacterial or viral antigens. The activated complex will promote the transcription of multiple pro-inflammatory cytokines (mainly IL-1b, IL-18) through activation of the Pro-caspase complex. It is well-known that HIV itself has binding sites for NF-kb, and it can participate in the activation from latent reservoirs.
During HIV infection, cellular signals to activate the inflammasome complex are produced (both in the GALT and the periphery), which will activate immune cells (creating new HIV targets and promoting viral persistence) along with an increase in IL1-b and IL-18.94 It has been seen that in "Immunological non-responders" (INRs) HIV patients there is an upregulation of the NLRP3 and caspase-1 pathway, which could explain the decrease in the CD4+ cell count (even on ART) through pyroptosis.105 Pyroptosis may be responsible, at least in part, for most of the CD4+ cell deaths during abortive replicative cycles.99 Importantly, pyroptosis is highly pro-inflammatory.
Inflammation may promote not only HIV persistence but also CD4+ cell loss. In HIV models, inflammasome activation was seen not only in monocyte-derived macrophages in gut tissue106 but also in liver macrophages (CD38+) where IL1-b was increased through the TLR4 – NLRP3 – Caspase pathway. As we see, there is a link between microbial translocation and inflammation, not only in the nearby mucosa and GALT but also in distant tissues like the liver.107 Interestingly, these highly infected IL1-b producing liver macrophages also switched to the M1 type or “Warburg Type” (see prior section) with a glycolytic pattern, promoting inflammation through exocytosis of vesicles in the nearby cells.
The Inflammasome complex is a highly conserved protein complex present in multiple tissues. Its activation in the Gut mucosa / GALT (Gut-Associated-Lymphoid-Tissue) will, in fact, amplify the inflammatory response in distant tissues, promoting inflammaging. Of note, cardiovascular disease, which has an increased incidence in HIV-positive patients, should be considered a chronic inflammatory disease.
4.1. Possible Mechanisms of InF to Overcome Inflammation and Cardiovascular Disease
For a more detailed description of the possible role of InF on preventing cardiovascular disease in HIV patients see our recent review (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8611195/).
It is important to clarify that even though InF has demonstrated many anti-inflammatory properties in animal studies, HIV patients present inflammatory levels far above the mean levels compared to HIV-negative controls, meaning that any decrease in systemic inflammation may correlate with much less organ damage and may decelerate the aging process significantly. InF has proven to be a very effective anti-inflammatory in multiple prior studies, mainly in animals, with conflicting results in humans [see references in the introduction section of108]. In rats exposed to InF and induced experimental stroke, decreased IL1-b, TNF-alpha, IL-6, and suppression of the “Inflammasome” were observed.109 InF could potentially “cool-down” the pro-inflammatory gut mucosa and decrease the TLR4 – NLRP3 – Caspase axis pathway, decreasing the immune activation of immune cells locally.
InF also resulted in reduced levels of mRNAs encoding the LPS receptor TLR4 and inducible nitric oxide synthase (iNOS) in the hippocampus of rats exposed to systemic LPS. Moreover, in another study, InF prevented the LPS-induced elevation of IL-1α, IL-1b, IFN-γ, RANTES, TNF-α, and IL-6.110 These two studies could have implications for decreasing the LPS-driven activation of TLRs, and therefore, gut and systemic inflammation. InF could also have an impact on impairing TLRs activation in distant tissues due to the fact that " leaked LPS " travels through the bloodstream to distant organs to activate inflammasomes locally (for example, in the atheromatous plaque in coronary arteries). Of note, LPS has an extremely high volume of distribution due to its lipid structure and could easily deliver the “spark” for activation of the Inflammasome in distant organs. These effects were recently tested in a proof-of-concept study in pre-diabetes patients exposed to InF.108 In this study, IL-6, hs-CRP, and TNF-alpha did not change, but InF decreased markers of oxidative stress, among other health benefits, even when the number of patients was low (n=8). If we consider inflammation a subtype of physiological stress, InF could help to cope with it, triggering adaptive cellular responses, counteracting local disease processes, decreasing DNA damage, promoting local tissue repair due to oxidative stress (see above stem cells), and increasing apoptosis of certain cells and cellular growth in others.48
It is now clear that Cardiovascular Disease (CVD) is considered a chronic inflammatory condition with the recruitment of activated monocytes and the creation of local inflammation in the atheromatous plaque with proliferation of “foamy cells.” It is well-known that HIV patients on ART suffer from severe cardiovascular disease compared to matched HIV-negative subjects. Trimethylamine N-oxide (TMAO) is an amine oxide produced in humans by intestinal microbiota from excess trimethylamine (TMA), an intermediate of choline metabolism. It has been linked to increased inflammation in adipose tissue and accelerates atherosclerosis.111 A decreased fasting mean level of 14.3 ng of TMAO versus a baseline mean of 27.1 ng (p = 0.019) was found in an InF study in humans,112 suggesting that InF can have implications for decreasing inflammation in the atheromatous plaque, not only by decreasing the recruitment of activated monocytes but by decreasing TMAO levels as well.
Theoretically, InF may provide cardiovascular protection in HIV patients on ART since CD14’s is a known marker associated with cardiovascular risk. Following the same line of thought, InF may also inhibit the development of the atheroma plaque in HIV patients by reducing the concentration of inflammatory markers, such as IL-6, homocysteine, and CRP, and at the same time, decreasing the migration of immune cells to the subendothelial area through the increase of Adiponectin.113 The decrease in the migration of inflammatory cells would be due to the decrease in the expression of the Vascular Cell Adhesion Molecule 1 (VCAM-1), Endothelial-leukocyte Adhesion Molecule 1 (ELAM-1), and Intracellular Adhesive Molecule 1 (ICAM-1) on vascular endothelial cells.113 It has also been seen that TMAO promoted vascular inflammation by activating the NLRP3 inflammasome locally, mediated in part through the inhibition of the SIRT3-SOD2-mitochondrial ROS signaling pathway, as we discussed before.
Interestingly, Proteobacteria was identified as one of the main producers of TMAO, which is increased in the dysbiosis caused by HIV.111 The above studies may be indirect evidence that InF may cause a reversal of the HIV-associated-dysbiosis with a decrease in Proteobacterias (mainly inflammatory and pro-glycolytic) with a switch to a healthier microbiome (with less production of TMAO) like Lactobacillus and Firmicutes (see under “Dysbiosis”). Recently, it was also seen that InF regulates the NLRP3 Inflammasome activation through SIRT1 (deacetylases), having implications as a therapeutic target.114 We reviewed in detail the important role of the Inflammasome for HIV persistence and inflammaging (see above).
InF decreased markers of liver inflammation significantly in experimental rodent models fed with pro-inflammatory diets,49 possibly through inhibition of TLR4, NF-kb, and interleukin signals.115 This could imply less activation of liver macrophages (like Kupffer cells), which feed off leaked microbial products through the portal circulation in HIV patients. Of note, immune-activated liver macrophages (M1 phenotype) are usually highly pro-inflammatory (see above). Decreasing liver inflammation and immune activation due to leaked products may be extremely important in order to protect liver cells from drug-induced hepatotoxicity or co-infections like HBV or HCV. This protective effect, if translated into humans, may have implications for protecting against fatty liver / NASH.116
Recent studies have shown that isocaloric TRF (Time Restricted Feeding) over 8 weeks in males reduced several markers of inflammation, including tumor necrosis factor alpha, interleukin 6, and interleukin 1b, while increasing adiponectin (an anti-inflammatory cytokine).117 It’s important to consider that this study involved healthy individuals with likely low levels of systemic inflammation, so the impact of InF on HIV patients on ART could be even more significant.
A study in mice demonstrated that enhancing mitochondrial energetics against damage-associated molecular patterns (DAMPs) may play a crucial role in preventing inflammation during fasting. This effect may be mediated, at least partially, through SIRT3-directed blunting of NLRP3 inflammasome assembly and activation.118 This finding could be important in HIV pathogenesis, considering that there may be some degree of mitochondrial damage due to long-term use of ART (mainly NRTIs), and that InF may, in part, restore mitochondrial bioenergetic functions and resistance to stress.
It has been proven that the ketone bodies β-hydroxybutyrate (BHB) and acetoacetate, both elevated during starvation, inhibit the NLRP3 inflammasome. As we discussed previously, InF significantly increases the concentration of SCFAs. The inhibitory mechanism is not solely explained by starvation-regulated mechanisms, such as AMP-activated protein kinase (AMPK), reactive oxygen species (ROS), autophagy, or glycolytic inhibition. SCFAs can inhibit the inflammasome by themselves, probably due to their high volume of distribution and high cell penetration. As a result, SCFAs have been shown to reduce NLRP3 inflammasome-mediated interleukin (IL)-1β and IL-18 production in human monocytes.119
A stressed Endoplasmic Reticulum (sER) is known to generate ROS, which, in turn, activates the NLRP3 inflammasome and secretion of IL-1b and IL-18. A recent study in rats revealed a potential therapeutic role for β-hydroxybutyrate in suppressing sER-induced inflammasome activation.120 In fact, InF may offer multiple benefits for other intracellular organelles in addition to its well-known effects on mitochondrial energetics. This could potentially regulate the intracellular trafficking and processing of HIV proteins, damaged DNA, or damaged proteins, in addition to increasing autophagy.
In another experimental model in rats with an experimentally induced stroke, InF attenuated the inflammatory response and tissue damage by suppressing NLRP1 and NLRP3 inflammasome activity in neural tissue.121 Protecting neural tissue and improving cognitive functions could be extremely important for the prevention of HIV-Associated Neurological Dysfunction (HAND) or other HIV-associated neurological syndromes (see below under “Cognitive Impairment”).
A revealing study demonstrated that patients with Rheumatoid Arthritis (RA) experienced significant clinical improvement (pain and inflammation) after a period of fasting if a vegetarian diet was followed thereafter.122 Another study of overweight Asthmatic female patients exposed to InF showed a significant decrease in the levels of TNF-alpha and markers of oxidative stress (8-isoprostane, nitrotyrosine, protein carbonyls, and 4-hydroxynonenal adducts), along with improved clinical response. Yet another study revealed that prolonged fasting blunted the NLRP3 inflammasome and Th2 cell activation in steroid-naïve asthmatics, as well as diminishing airway epithelial cell cytokine production.123
These two human studies highlight the possibility of applying InF to counteract chronic inflammatory conditions, which, in turn, promote accelerated aging. In fact, HIV is a perfect example of a chronic inflammatory disease. We believe that in all these conditions (RA, Asthma, and HIV), the baseline level of inflammation is so high that any degree of decrease may have significant clinical implications. These two studies showed not only that it is feasible to use this ancient adaptive cellular mechanism on a daily basis to fight inflammation in human subjects, but also that it is possible to manipulate inflammation using nutritional strategies. As an example, a diet rich in carbohydrates and fat can increase the levels of insulin and leptin. Leptin usually reflects a “pro-inflammatory state”, whereas adiponectin and ghrelin (both increased while fasting) can suppress inflammation and increase insulin sensitivity. Adiponectin also possesses anti-atherosclerotic and anti-inflammatory effects by inhibiting the adhesion of monocytes to endothelial cells (see above).
5. Indoleamine 2,3-dioxygenase 1 (IDO-1)
For more details about the role of IDO-1 on HIV pathogenesis see our recent review: (https://nortonhealthcaremedicaljournal.scholasticahq.com/article/118564-unveiling-the-nexus-of-cd38-overactivation-nad-depletion-and-mitochondrial-dysfunction-in-immunological-failure-among-virologically-suppressed-hiv).
IDO-1 is believed to play a central role in HIV pathogenesis through its immunoregulatory effects, which have been extensively reviewed elsewhere.61–63,65,66,78,79,97,98 This enzyme transforms Tryptophan to N-formyl-D-kynurenine and increases the Kynurenine/Tryptophan ratio. IDO-1 also promotes the conversion of naïve T cells to T.regs,97 which is detrimental in HIV/SIV immunopathogenesis (see above). There is an increase in IDO-1 activity in some difficult-to-reach sites like the testicles, eyes, and brain, indicating that it may also be involved in HIV persistence in “sanctuary sites”. There is a direct relationship between increased IDO-1 activity and increased IL-1b, IL-6, IL-18, IP-10, TNF-a, sCD40L, LPS, and sCD14.99 An extensive review of the literature showed that this enzyme is the focus of multiple clinical trials as a possible therapeutic target, even in cancer research.
LPS, HIV-1 Tat protein,124 IFN-alpha/gamma, Aryl Hydrocarbon Receptor (AHR) activation, Toll-like receptors, and loss of Th17 cells are among the promoters of this enzyme’s activity. Increased IDO-1 activity has been linked to increased pro-viral DNA, increased viral reservoir, poor CD4+ recovery, HIV progression, AIDS and non-AIDS events, and increased mortality.62,97
After 2 years of effective ART, HIV patients with high N-formyl-D-kynurenine in plasma had higher CD14’s, a higher percentage of T.regs, a lower percentage of naïve cells, a lower CD4/CD8 ratio, a decreased amount of Th17 cells,68 and poor immune reconstitution compared to patients with low kynurenines.125
A recent study also showed a correlation between IDO-1 activity and HIV DNA in blood (reflecting its possible influence in the reservoir size), immune activation, and T cell exhaustion.97
In human monocytes derived cells (MoDC), HIV-1 Tat protein induced IDO-1 expression and activity in an NF-κB-dependent manner by recruiting the TLR4 pathway.124
Importantly, immune exhaustion in CD4+ infected cells is associated with higher viral loads, decreased replicative capacity, and deficient cytokine responses.68 The dysbiosis present in pathogenic SIV/HIV models with depletion of Lactobacillus and an increase in Proteobacteria and Prevotella (see below under “Dysbiosis”) could also promote IDO-1 activity. It has been observed that Lactobacillus supplementation decreased IDO-1 activity in SIV models,126 and dysbiosis.68 In fact, the supplementation of IL-22 along with probiotics could decrease microbial translocation and inflammation, ultimately decreasing IDO-1 activity (see below).71
In Figure 4 can be seen the Kynurenine pathway with some of its intermediate metabolites and their physiologic effects:

On Figure 5 can be seen the possible relationship between IDO-1, CD38(+), NAD(+) depletion, HIV-Associated Mitochondrial Dysfunction (HIVAMD), Immune-activation, chronic inflammation, Immune-senescence, and Aging in PLWH. For a more detailed explanation explore our recent review on this journal (https://nortonhealthcaremedicaljournal.scholasticahq.com/article/118564-unveiling-the-nexus-of-cd38-overactivation-nad-depletion-and-mitochondrial-dysfunction-in-immunological-failure-among-virologically-suppressed-hiv):
,_HIVAMD,_Immune-activation,_Chronic_inf)
Summary of IDO-1 role in HIV pathogenesis based on an extensive literature review105,106,127,128:
-
LPS-stimulated-TLR4 in APCs will increase the activity of IDO-1, which transforms Tryptophan to N-formyl-D-kynurenine and increases the Kynurenine/Tryptophan ratio.
-
The increased Treg/Th17 ratio will also increase IDO-1 activity.65
-
IDO-1 will stimulate the NF-kb factor, increasing pro-inflammatory cytokines.
-
There will also be an increment of glycolysis products, favoring the switch to the M1 (pro-inflammatory) phenotype in macrophages and APCs (see above section).
-
N-formyl-D-kynurenine also inhibits TH17 cells, which, through the secretion of IL-17, play a primary role in tight junction homeostasis and repair (increasing the production of tight junction proteins Claudin 1-2).
-
Kynurenine will also promote the formation of T.regs (FOXp3 – CD39 phenotype), which decreases the T-lymphocyte cytotoxic cell response and promotes HIV persistence. In the GALT (Gut Associated Lymphoid Tissue), the increased amount of Tregs promotes the formation of TGF-B1, which, in turn, increases the transcription of Collagen mRNA (promoting GALT fibrosis), decreases T cell function, and increases apoptosis.
-
The resulting abnormal architecture of the GALT tissue promotes even further lack of HIV clearance (and also probably co-pathogens) and decreases local immune function in general.
-
Unfortunately, the increase in Tregs and IDO-1 activity, along with GALT fibrosis, persists even while on effective ART.79
-
N-formyl-D-kynurenine may also stimulate PD-1 receptors on the surface of CD4+T cells, favoring the activation of mTOR and promoting HIV persistence since data suggest that PD-1 negatively regulates T cell responses.
5.1. Possible Mechanisms of InF to Overcome IDO-1
-
InF could change or reverse the HIV-associated-dysbiosis (predominance of Proteobacterias) with less generation of “Indoles” (see below under “Indoles”), as Indols activates the Aryl Hydrocarbon Receptor (AHR), which, in turn, will activate IDO-1.
-
There will be less pro-inflammatory cytokines locally with less activation of the IDO-1 pathway since inflammatory mediators trigger its activity (see above under “inflammation”).
-
InF may decrease microbial translocation (LPS) with less inflammation caused by APCs due to potential repair of the tight junctions defect.
-
InF may may cause the Kynurenine/Tryptophan ratio to approach 1 (reversal of the ratio) as seen in the clinical trial below (see 5.2 below)
-
InF may cause a decrease in the immune metabolic switch of Macrophages and APCs to the “Warburg-like Phenotype” (see above) with less further activation of IDO-1.
-
The inhibition of IDO-1 activity with InF may have some impact on the HIV reservoir size (not only in the gut mucosa) and on the CD4+/PD-1 cells (it is believed that kynurenines activate the mTOR in those cells through PD-1 mediated pathways and promote persistency and latency).
5.2. Prolonged Fasting can Alter Kynurenine Intermediate Metabolites
A very recent revealing study (https://www.nature.com/articles/s41430-024-01451-7.pdf) explores how prolonged fasting (6 days) impacts kynurenine pathway metabolites and stress markers in healthy males. Twenty four healthy males aged 18-44 with BMI 19.5-29.9 kg/m² were divided into fasting (FAST, n=14) and control (CON, n=10) groups. The FAST group followed a zero-calorie diet for 6 days with water ad libitum, while the CON group maintained usual diets. Blood and saliva samples were collected at baseline, Day 2, Day 4, Day 6, and one week post-fasting. Plasma levels of tryptophan (TRP), kynurenine (KYN), kynurenic acid (KYNA), 3-hydroxykynurenine (3-HK), picolinic acid (PIC), quinolinic acid (QUIN), and nicotinamide (NAM) were measured. Stress markers including cortisol, adrenaline, and noradrenaline were also analyzed.
It was documented that prolonged fasting significantly increased KYNA, 3-HK, and Picolinic acid levels, peaking at Day 6. TRP and Quinolonic acid levels decreased, while KYN and NAM remained stable. Cortisol and noradrenaline levels showed no significant changes, but adrenaline levels peaked on Day 4 in the FAST group. All kynurenine pathway metabolites returned to baseline levels after resuming regular diet, except for KYNA which remained elevated. The study indicates that prolonged fasting activates the kynurenine pathway, affecting TRP metabolism and increasing several metabolites involved in immune modulation and energy production. These changes are largely reversible upon refeeding, suggesting metabolic adaptability. The lack of significant changes in stress markers suggests minimal stress impact from fasting. The findings highlight the potential of fasting to modulate metabolic pathways implicated in various diseases.
Implications for HIV Patients
The kynurenine pathway is known to be accelerated in HIV patients (see above), contributing to chronic inflammation, immune dysregulation, and neurocognitive disorders. The findings from this study suggest several potential implications for HIV patients:
-
Modulation of Immune Response: Increased levels of Kynurenic acid (KYNA) and Picolinic acid (PIC), observed during fasting, might help modulate immune responses in HIV patients. KYNA has neuroprotective and anti-inflammatory properties, which could be beneficial in managing HIV-associated neurocognitive disorders (HAND) and reducing chronic inflammation.
-
Reduction of Chronic Inflammation: Fasting-induced changes in the kynurenine pathway metabolites may help reduce the systemic inflammation seen in HIV patients. For instance, decreasing TRP and increasing anti-inflammatory metabolites like KYNA and PIC could mitigate the inflammatory processes exacerbated by HIV.
-
Improvement in Energy Metabolism: The study’s findings that fasting boosts metabolites like PIC, which are involved in energy production, might help address the energy deficits and fatigue commonly experienced by HIV patients.
-
The decrease of Quinolonic acid (QA) is concerning due to the fact that it represents an important precursor of NAD(+) generation; “de-novo pathway” (through the activity of the Quinolate Phosphoribosyl transferase – QPRT -). At the same time the decrease of QA is neuroprotective. Since there are 2 other alternative ways to generate NAD(+) [ from Nicotinic acid and the “Salvage pathway”) it may not be concerning at all.
-
The increased levels of 3-HK may also be counterproductive since may cause HIVAMD
-
Potential Therapeutic Strategy: Incorporating prolonged fasting or intermittent fasting (InF) regimens could be explored as a non-pharmacological strategy to modulate the kynurenine pathway, potentially improving immune function and reducing HIV-associated comorbidities.
-
Further Research Needed: While the study shows promising results, further research is needed to evaluate the safety, feasibility, and long-term effects of fasting in HIV patients, particularly those on ART. Clinical trials should be conducted to determine the optimal fasting protocols and assess their impact on the kynurenine pathway and overall health outcomes in HIV patients.
6. Microbiome alteration (Dysbiosis)
An extensive review of the literature points towards a profound alteration of the gut microbiome in HIV patients compared to HIV-negative controls, even while on effective ART.61–63,65,66,78,98,129 In HIV patients, there is a significant increase in Proteobacteria, Candida (probably with increased translocation of BDG), and Prevotella with a decrease in Bacteroides, Firmicutes (importantly, Roseburia), Bifidobacterium, and Lactobacillus.
The increase in Proteobacteria is correlated with heightened T-cell activation in the blood and gut, lower mucosal IL-17/IL-22 secretion, and a higher plasma N-formyl-D-kynurenine/tryptophan ratio (see above).65 Usually, an increase in Proteobacteria translates into gut damage and inflammation.78 Of note, even some ARVs like Protease Inhibitors (PIs) may show some deleterious effects against protective flora as well.63 Interestingly, Bacteroides and Firmicutes play an important role in the secretion of SCFAs like Propionate, Butyric acid, Acetate, and Valeric acid. Importantly, Roseburia (Firmicutes), which is reduced in HIV dysbiosis, plays an important role in the regulation of the immune system, Treg regulation, and an increase in Bacteriocin-like substances (BLIS).78 Lachnospiraceae, also decreased in HIV patients, has been linked to protection against colon cancer due to its production of Butyric acid.78
It remains to be fully elucidated which phenomena occur first:
-
Dysbiosis
-
Decreased SCFAs
-
Microbial translocation.
Some proteomic analysis suggests that the metabolic changes (decreased SCFAs) actually occur before the structural epithelial damage, which may imply that the microbiome change precedes the tight junction lesions (?).78 Other evidence (reviewed above) shows instead that the structural lesions occur as early as 3 days after SIV infection.
6.1. Possible Mechanisms of InF to Overcome HIV-Associated Dysbiosis
-
InF could promote a shift towards a healthier microbiome130 as documented during Ramadan-associated intermittent fasting. See also a recent review:(https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8670288/).
-
The predominantly “diurnal” feeding patterns (early time restrictive feeding – eTRF -) that some InF regimens promotes, along with prolonged periods of fasting (at least 16-18 hours/day on average for different modalities), could promote less gut inflammation, promote bacterial diversity (with a decrease in the predominance of Proteobacteria and Prevotella),
-
Decrease microbial translocation, with less local/systemic inflammation/immune activation, potentially due to tight junction repair
-
Decrease Indols from Proteobacteria
-
Increase Lachnospiraceae with increased Butyric acid130
-
Decrease post-prandial endotoxemia. Overfeeding has been associated with an increase in endotoxemia (“leaky-gut”) with inflammation and an increase in CD14’s.131
-
InF, along with probiotic supplementation, could potentially restore some of the lost Lactobacillus, Bacteroides, and Firmicutes (mainly Roseburia), as these species contribute to the production of SCFAs instead of Indole-producing species like Proteobacteria.
7. Short Chain Fatty Acids (SCFAs)
As we reviewed in prior sections, SCFAs will be notably increased during InF periods. Of note, Butyrate is particularly important for colonic health because it is the primary energy source for colonocytes. Butyrate may have implications for epithelial barrier repair and integrity after the initial HIV/SIV structural damage. SCFAs help boost the protective mucus layer in the gut and also have the ability to influence genes that control cell proliferation and cell cycles. SCFAs can increase the secretion of antimicrobial peptides and the secretion of IL-18 by epithelial cells, helping to maintain the anatomical and functional integrity of the epithelial barrier.78 SCFAs’ primordial role in gut homeostasis was recently revealed in a trial on rats where exposure to Ceftriaxone promoted intense dysbiosis with a significant decrease in SCFAs, along with an increase in gut leakage, and inflammatory cytokines (TNF and IL-10). It was also found that circulating levels of butyric acid are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis.132 In an inflammatory environment (like HIV/SIV), it has been shown that SCFAs can decrease IL-12 and TNF-alpha.78 Adding to its multiple properties, it has been observed that Butyric acid and Acetoacetate can also decrease inflammation through inhibition of the NLRP3 inflammasome-mediated inflammatory response with significant reductions of interleukin (IL)-1β and IL-18 production in human monocytes (see above under “Inflammation”).119
It has also been confirmed that SCFAs (mainly Butyric acid) inhibit the activity of histone deacetylase (HDAC) [an enzyme involved in post-translational modifications, namely the process of deacetylation] and the process of histone crotonylation.133 Histone deacetylase inhibitors (HDACi) are being evaluated in a “shock-and-kill” or “Purge” therapeutic approach to reverse HIV-1 latency from CD4(+) T cells and other reservoirs. HDACi could induce HIV RNA synthesis in latently infected cells and facilitate eradication after being targeted with ART. Therefore, in addition to known HDACi compounds like belinostat, givinostat, panobinostat, romidepsin, and vorinostat, Butyric acid could play that role as well. Intriguingly, the above mechanism involved unc-51-like autophagy-activating kinase 1 (ULK1) and the inhibition of the mammalian target of rapamycin (mTOR) (see above under “mTOR”) inhibited by InF. Mechanistically, it requires the formation of autophagosomes and their maturation into autolysosomes providing evidence in support of a link between HDACi, mTOR, and autophagy, potentially as a valuable tool for HIV control.134 The use of InF as HDACi remains hypothetical and needs further exploration.
As we discussed previously, HIV-1 induces the first signal to activate the NLRP3 inflammasome in monocyte-derived macrophages [probably via TLR8-mediated mechanisms and activated caspase-1135] with a posterior increase in IL1-b.106 Upregulation of NLRP3 and caspase-1 was also seen in immunologically non-responders (INRs). Caspase-1 upregulation could induce CD4 T-cell loss via “Pyroptosis”.105 The increase in Butyric acid seen with prolonged ketosis (“days of food restriction”) could potentially contribute to the reversal of immunological failure to ART seen in these cases if the NLP3 – Caspase-1- Pyroptosis pathway is interrupted (hypothetical).
7.1. Possible Mechanisms of InF to Increase the Production of SCFAs
-
InF will significantly increase the production of SCFAs. However, longer fasting periods lead to higher peak levels of ketone bodies, resulting in a more intense state of ketosis."
-
The reversal of the dysbiosis will switch the microbiome to a one prone to produce SCFAs
8. Indoles
Another critical aspect in the pathogenesis of chronic HIV and gut inflammation is the increased production of Indoles by the overgrowth of Proteobacterias during the HIV-induced dysbiosis. Indoles are a derivative of the amino acid tryptophan. It is well-known that Proteobacterias promote a pro-inflammatory environment (see above).136 One of the consequences of the excess of Indole groups is the overactivation of the aryl hydrocarbon receptor (AHR), which, in turn, stimulates IDO-1, with all the consequences explained previously (see above under “IDO-1”). It was seen that AHR signaling, via IL-22, inhibits inflammation and colitis in the gastrointestinal tract of mice, which could be beneficial for epithelial integrity in SIV/HIV pathogenesis.137 Of note, AHR may also inhibit TH17 cells (at least in a model of allergic rhinitis), which could be detrimental for epithelial integrity.138 As we can see, AHR activation could have beneficial and detrimental consequences for HIV pathogenesis, with its most significant detrimental consequence being the activation of IDO-1 with increased Tryptophan to N-formyl-D-kynurenine and its immunomodulatory effects.
8.1. Possible Mechanisms of InF to Decrease Indol Production
-
InF could potentially reverse the predominance of Proteobacteria with a significant decrease in the production of Indoles (see above under “Dysbiosis”). See: (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7029019/).
-
InF could potentially inhibit or decrease the detrimental activation of IDO-1 and AHR by indoles, decreasing the Kynurenine/Tryptophan ratio.
-
Nutritional manipulation along with prebiotics/probiotics, may also have a negative impact on the generation of Indoles, along with a decrease in Proteobacteria and decreased IDO-1 activity.
9. HIV Associated Neurocognitive Disorders (HAND)
HIV-associated neurocognitive disorders (HAND) are a heterogeneous group of diseases that remain prevalent even in virologically suppressed HIV patients on ART. HAND can be divided into three categories:
-
HIV-associated asymptomatic neurocognitive impairment (ANI)
-
HIV-associated mild neurocognitive disorder (MND)
-
HIV-associated dementia (HAD).139–143
The etiology of these diseases is multifactorial, and many confounders (aging, depression, drug abuse, opportunistic CNS disease, and co-infection with HCV) contribute to their pathogenesis. Like in other models of tissue damage during HIV/SIV infection, there is evidence showing an association between microbial translocation, monocyte activation, and neuronal damage,144–146 supporting the hypothesis of distal organ damage from the GI tract (Gut-Brain axis).
It is possible to measure the degree of cognitive impairment and classify these disorders through neurocognitive tests as well as measuring some markers of inflammation and monocyte activation:
-
Direct methods: Neurocognitive tests, MRI, PET scans, Amyloid scans.
-
Indirect methods: IL-6,144–146 sCD14,144 sCD163,144 LPS,145 CXCL-10.
“…We view HAND as just one more part of the spectrum of consequences of chronic inflammation and immune activation in distant organs…..”
Brain aging predisposes to Alzheimer’s, Parkinson’s, and Huntington disease.147 Chronic Inflammation triggers insulin resistance, which accelerates neurodegeneration. Activation of the pro-inflammatory factor NF-kb in the hypothalamus also promotes neuronal aging. Brain aging also decreases synaptic plasticity, which is essential for learning and memory storage.147 A decline in neurogenesis due to inflammation and senescence may also be responsible for HAND.147 A loss of mitochondrial effectiveness and bioenergetics, with an increase in ROS, is found in senescent tissues and also plays a role in neurodegeneration. In fact, damaged mitochondria activate apoptotic programs with cell death, as we discussed above with the NLRP3-ASC-Pro caspase-1 pathway with “pyroptosis”.147
There is ample evidence that inflammation plays an important role in HAND.139–144,148 Both inflammation and oxidative stress are responsible for neurotoxicity in the context of HIV-1-associated dementia (HAD).99 As proof of the above a recent study showed that exposure of human primary microglia to ssRNA40 (HIV single-stranded RNA) activates the (NLRP3) inflammasome with increased secretion of the pro-inflammatory cytokines IL-1β, IL-18, and neurotoxic cytokines TNF-α and IL-1α.118 In another recent study in which HIV-Tg rats were administered LPS, it was shown that HIV-1 Tat activates microglial NLRP3 Inflammasome-Mediated Neuroinflammation.127
“….This is another proof of the “Gut-Brain axis”, as it showed how gut immune activation and microbial translocation can potentially create cognitive impairment through inflammatory signals….”
9.1. Possible Mechanisms of InF to Overcome HAND
-
Interestingly, a recent study revealed a “Brain-Gut pathway” that maintains homeostasis during fasting periods through its effects on epithelial integrity, increasing epithelial signals to strengthen tight junctions and potentially decreasing translocation.149
-
It was shown in animals that InF attenuated the cognitive impairment and cerebral oxidative stress caused by systemic inflammation, resulting from a decrease in LPS in plasma.110 In fact, the increased IDO-1 activity (see above) detected during HIV/SIV-induced dysbiosis could contribute to HAND,63 highlighting the importance of controlling gut inflammation to improve cognitive functioning.
-
Multiple neuronal benefits have been demonstrated in animals exposed to InF, including increased neuronal plasticity, promoted neurogenesis from stem cells, and improved learning and memory.
-
It is well-known that InF increases alertness/arousal and mental acuity, possibly as an evolutionary response in the context of food scarcity.50 The increased cellular stress and challenge that starvation signifies to neurons triggers these archaic mechanistic pathways, directed towards becoming more resistant to stress or more prone to looking for food in the environment in order to survive.
-
The prior neuronal protective mechanisms were challenged in a study of rats where those exposed to InF for 3 months were more resistant to ischemic-induced damage than rats fed ad libitum.11,50,109
-
It has also been shown that starvation increases brain-derived neurotropic factor (BNDF).48 BDNF promotes the growth and maintenance of synapses and dendrites, and increases the de novo production of neurons from stem cells.
-
InF increases mitochondrial bioenergetics and stress resistance through an increase in BDNF, PGC-1alpha (genes of mitochondrial bioenergetics), and Sirtuin 3 (suppresses oxidative stress and apoptosis).50
-
Notably, rats were protected while fasting against neuronal degeneration and dysfunction in models of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease. In neurodegenerative diseases, it is clear that autophagy is impaired, likely due to the overactivation of mTOR.91
-
InF attenuated the neuroinflammation of hypothalamic and hippocampal neurons of rats exposed to LPS. The neuronal protection was thought to be through inhibition of the NF-kb factor, reduced pro-inflammatory cytokines (in blood and brain), higher expression of BDNF, and preserved spatial memory.147
-
Another recent study in rats showed that induction of neuronal autophagy in an experimental model of Huntington’s disease was achieved by intermittent fasting, an intervention known to inhibit mTOR.150
-
For centuries, the “anti-epileptic properties of starvation” have been known, mainly due to a switch to a predominantly ketone-based neuronal metabolism.151 Actually, centuries ago, patients with seizures thought to be “possessed by demons” were locked for days without eating for treatment, with subsequent clinical improvement. Ketone body metabolism reduces ROS toxicity in tissues through the NADPH system.152
-
When fasting rats were exposed to the neurotoxin agent Kainic acid, their hippocampal neurons showed increased survival, with preserved learning and memory compared to rats fed ad libitum.93
Some of the general mechanisms of its neuroprotective effects are:
-
Decrease systemic inflammation (TNF-alpha, IL1-b, and IL-6)
-
Decrease neuronal inflammation
-
Decrease accumulation of oxidatively damaged molecules (oxidative stress)
-
Improved cellular mitochondrial bioenergetics
-
Stimulate neurogenesis from stem cells
-
Increase neural plasticity
-
Decreased neuronal DNA damage
-
Increase neuronal-glial autophagy (through inhibition of mTOR92,153,154 similar to Rapamycin)
-
Increase protein chaperones (HSP70 and GRP78)
-
Increase in BNDF and FGF2.50,109,155
10. Metabolic Complications of PLWH
For a more detailed description of the metabolic consequences of HIV please see our recent review on: (https://nortonhealthcaremedicaljournal.scholasticahq.com/article/118562-is-hiv-associated-mitochondrial-dysfunction-hivamd-related-to-weight-gain-and-metabolic-complications-on-plwh-contribution-of-ultra-processed-foods).

10.1. How InF can address most of the metabolic complications of PLWH
Addressing the Core Issues: Inflammation, Microbial Translocation, and Mitochondrial Dysfunction
-
Decreasing Inflammation: InF has demonstrated significant anti-inflammatory effects in both animal studies and, to a lesser extent, in humans. As described in our previous review, the primary driver of metabolic abnormalities in HIV is chronic inflammation. By reducing inflammation, InF could potentially:
-
Reduce Insulin Resistance: Inflammation can interfere with insulin signaling pathways, leading to insulin resistance. InF’s anti-inflammatory effects could potentially improve insulin sensitivity, allowing cells to use glucose more effectively.
-
Reduce Fat Accumulation: Inflammation promotes fat storage, especially in the liver and visceral areas, contributing to fatty liver and weight gain. Reducing inflammation could potentially reverse this process. Also, lipolysis, which is promoted by InF could decrease the fat mass as well.
-
Modulate the Kynurenine Pathway: InF can influence the kynurenine pathway, a metabolic pathway closely linked to inflammation. This pathway can generate pro-inflammatory metabolites that can contribute to metabolic complications. Modulating this pathway through InF could potentially reduce inflammatory load.
-
-
Reducing Microbial Translocation: Microbial translocation, the leakage of bacteria and their products from the gut into the bloodstream, is a major contributor to inflammation and metabolic complications in HIV. InF could potentially improve gut health by:
-
Restoring Gut Microbiome Balance: InF can promote a healthier gut microbiome, reducing the dominance of pro-inflammatory bacteria like Proteobacteria and increasing beneficial bacteria like Firmicutes and Lactobacillus. This could decrease microbial translocation and inflammation.
-
Strengthening the Gut Barrier: InF can help repair the damaged gut lining by promoting the production of SCFAs (short-chain fatty acids), which are crucial for gut barrier integrity. This could reduce the leakage of microbial products into the bloodstream.
-
-
Improving Mitochondrial Function: Mitochondrial dysfunction (HIVAMD) is a key factor in metabolic complications in HIV. For a more detailed description of the contribution of HIVAMD to the aging process on PLWH see our recent review on: (https://nortonhealthcaremedicaljournal.scholasticahq.com/article/117234-accelerated-aging-process-in-patients-living-with-hiv-role-of-mitochondrial-dysfunction).InF can potentially improve mitochondrial health by:
-
Enhancing Energy Production: InF promotes metabolic adaptation, shifting the body’s energy production towards fatty acid oxidation (ketogenesis) rather than glucose reliance. This can reduce strain on mitochondria and enhance their efficiency.
-
Reducing Oxidative Stress: InF can decrease oxidative stress, a major contributor to mitochondrial damage. This can protect mitochondria from further deterioration.
-
Promoting Autophagy: InF can stimulate mitophagy, a cellular process that removes damaged mitochondria. This helps to clear out dysfunctional mitochondria and promote the growth of healthier ones.
-
-
Addressing Specific Metabolic Abnormalities:
-
Fatty Liver: InF has shown promise in reducing fatty liver in both animal models and humans, likely due to its effects on reducing inflammation, improving insulin sensitivity, and promoting fat burning (lipolysis).
-
Diabetes: InF has been demonstrated to improve insulin sensitivity and blood glucose control in individuals with pre-diabetes and type 2 diabetes. In HIV, InF could potentially help manage or even prevent the development of diabetes, particularly in those with insulin resistance.
-
Hyperinsulinemia: By improving insulin sensitivity, InF could potentially reduce the need for the pancreas to overproduce insulin, leading to a decrease in hyperinsulinemia.
-
Weight Gain: InF can promote weight loss by reducing appetite, improving insulin sensitivity, and increasing energy expenditure. This could help manage weight gain in PLWH on ART.
-
11. Recent studies reporting increased cardiovascular risk with Intermittent fasting
A recent online publication by the American Heart Association (AHA) raised eyebrows worldwide when they reported that 8-hour time-restricted eating linked to a 91% higher risk of cardiovascular death (https://newsroom.heart.org/news/8-hour-time-restricted-eating-linked-to-a-91-higher-risk-of-cardiovascular-death). They reviewed information about dietary patterns for 20,000 participants in the annual 2003-2018 National Health and Nutrition Examination Surveys (NHANES) in comparison to data about people who died in the U.S., from 2003 through December 2019, from the Centers for Disease Control and Prevention’s National Death Index database. After a waive of global criticism worldwide the authors mentioned, quote: “Although the study identified an association between an 8-hour eating window and cardiovascular death, this does not mean that time-restricted eating caused cardiovascular death.”
The study’s serious limitations included:
-
Retrospective nature
-
It was a POSTER PRESENTATION in a scientific meeting, not a peer reviewed
-
Its reliance on self-reported dietary information, which may be affected by participant’s memory or recall and may not accurately assess typical eating patterns.
-
Factors that may also play a role in health, outside of daily duration of eating and cause of death, were not included in the analysis. Was the group with the shortest time-restricted eating window unique compared to people who followed other regular eating schedules in terms of weight, stress, traditional cardiometabolic risk factors or other factors associated with adverse cardiovascular outcomes (?).
-
No information about the nutrient quality of the diets typical of the different subsets of participants. Without this information, it cannot be determined if nutrient density might be an alternate explanation to the findings that currently focus on the window of time for eating.
-
The categorization into the different windows of time-restricted eating was determined on the basis of just two days of dietary intake.
“….It is counterintuitive to think that any form of daily calorie restriction (including inF) could increase cardiovascular risk…”
Future well-designed, prospective studies will need to be performed to answer the question of a potential relationship between InF and cardiovascular disease.
Conclusion
While antiretroviral therapy (ART) has revolutionized HIV management, transitioning it from a fatal disease to a chronic condition, addressing the persistent inflammatory state that often accompanies HIV infection remains a critical challenge. This review explores the potential benefits of Intermittent Fasting (InF) as a non-pharmacological intervention for HIV patients, focusing on its ability to mitigate the multifaceted consequences of chronic inflammation, including metabolic dysregulation, gut dysbiosis, and neurocognitive decline. InF’s potential to promote a healthier gut microbiome, reduce microbial translocation, and strengthen the gut barrier suggests a significant impact on reducing chronic inflammation and its systemic effects. By shifting the body’s metabolic pathways towards a more energy-efficient state, InF may improve mitochondrial function, enhance cellular stress resistance, and slow the accelerated aging processes often observed in HIV patients. In addition to its impact on gut health and mitochondrial function, InF may positively influence the kynurenine pathway, potentially reducing the levels of pro-inflammatory metabolites and enhancing the production of neuroprotective metabolites. This suggests a potential role for InF in mitigating HIV-associated neurocognitive disorders (HAND) and promoting neuronal health. Furthermore, InF’s ability to modulate the NLRP3 inflammasome and its associated inflammatory responses, along with its potential to enhance autophagy, underscores its diverse therapeutic potential.
While the research on InF is promising, particularly in animal models, it is essential to acknowledge that further investigation, including well-designed clinical trials in HIV patients, is necessary to confirm its safety, efficacy, and long-term benefits. However, the intricate interplay of InF with various aspects of HIV pathogenesis, from gut health to immune modulation and neuroprotection, makes it a compelling candidate for further exploration as a holistic intervention for people living with HIV.