
Introduction
With the advent of antiretroviral therapy (ART), HIV has transitioned into a chronic disease, offering patients a lifespan comparable to HIV-negative individuals.1–12 However, despite effective virologic suppression in plasma, around 30% of people living with HIV (PLWH) still undergo immunological failure, characterized by an inadequate CD4+ T cell recovery. These individuals, known as “Immunological non-responders” (INR), present a clinical enigma as the etiology behind this phenomenon remains largely undefined.13–19 Unraveling the pathogenesis of immunological failure is crucial, given that INRs suffer from higher morbidity and mortality rates compared to full responders20 Remarkably, several clinical interventions have been unsuccessful in reversing or improving this CD4 cell response deficit. INRs is generally defined as CD4+T cell count below 350 cells/μl at 2 years after ART initiation, with undetectable plasma HIV RNA.21
CD38: A Multifaceted Molecule with Diverse Functions
CD38 is a type II transmembrane glycoprotein which exists in two forms: Membrane-bound - Anchored to the cell surface - , and Soluble - Found circulating in bodily fluids -. CD38 is expressed on various immune cells, including B lymphocytes, T lymphocytes (activated), Natural killer (NK) cells, Monocytes, Macrophages, Dendritic cells, and Platelets.5
CD38 boasts a diverse range of functions including 5,22:
-
Receptor:
- Binds to CD31, influencing cell adhesion and migration.
-
Enzyme:
-
NADase: Breaks down NAD+ into ADPR and cyclic ADPR, impacting cellular NAD+ levels.
-
ADP-ribosyl cyclase: Catalyzes the formation of cyclic ADP-ribose from NAD+, involved in calcium signaling.
-
-
Role in Immune Response:
-
Contributes to immune cell activation, proliferation, and differentiation.
-
Plays a role in inflammatory processes.
-
-
Possible role on Chronic inflammatory diseases and Aging
NAD+ Explained: A Vital Molecule for Life
Nicotinamide adenine dinucleotide (NAD+) is a crucial coenzyme found in all living cells. It exists in two forms: NAD+ (oxidized) and NADH (reduced) .23 The molecule is constantly switching between these forms to carry out essential functions. NAD+ is found in every cell.
NAD+ plays a starring role in numerous cellular processes, including:
-
Energy metabolism: essential for optimal mitochondrial function
-
DNA repair: NAD+ helps enzymes repair damaged DNA, protecting against mutations and aging (cofactor of “Sirtuins”).
-
Gene expression: It influences gene activity, impacting various cellular functions.
-
Cell signaling: NAD+ participates in communication between cells, coordinating bodily processes.
-
NAD+ acts as a cofactor for several essential enzymes, including:
-
Sirtuins: Involved in DNA repair, metabolism, and longevity.
-
PARPs: Play a role in DNA repair and cell death.
-
CD38 and CD157: Enzymes involved in cell signaling and immune functions.
As we age, NAD+ levels naturally decrease. This decline is linked to various age-related issues, including14,23:
-
Metabolic disorders
-
Neurodegenerative diseases
-
Frailty
-
Accelerated aging
Mitochondrial Dysfunction: A Cellular Energy Crisis
Mitochondrial dysfunction refers to the impaired function of mitochondria, the powerhouses of our cells responsible for generating energy (ATP).12–14
Several components within the mitochondria can be affected, leading to dysfunction:
-
Electron transport chain (ETC): Responsible for the final stages of energy production. Defects here can significantly hamper ATP production mainly through impairment of mitochondrial oxidative phosphorylation (OXPHOS).
-
Mitochondrial DNA (mtDNA): Mutations or damage to mtDNA can disrupt energy production processes since many mitochondrial proteins are derived from mDNA.
-
Mitochondrial membrane: Damage to the membrane can affect the import of essential molecules and disrupt energy production affecting concentration gradients which usually drives ATP production.
-
Lack of essential enzymatic cofactors like NAD+
The consequences of mitochondrial dysfunction can be wide-ranging and severe, including24,25:
-
Fatigue and muscle weakness
-
Neurological problems: potentially contributing to neurodegenerative diseases.
-
Increased oxidative stress:
-
Metabolic disorders: Disrupted energy metabolism can lead to conditions like diabetes and obesity. Many common diseases are thought to be caused ultimately from some degree of mitochondrial dysfunction.
-
Accelerated Aging: mitochondrial dysfunction is one of the “Hallmarks of Aging”.
We called HIV-Associated Mitochondrial Dysfunction (HIVAMD) the mitochondrial dysfunction derived from the HIV itself or its treatment.
Etiology of Immunological Failure: When Uncertainty Reins
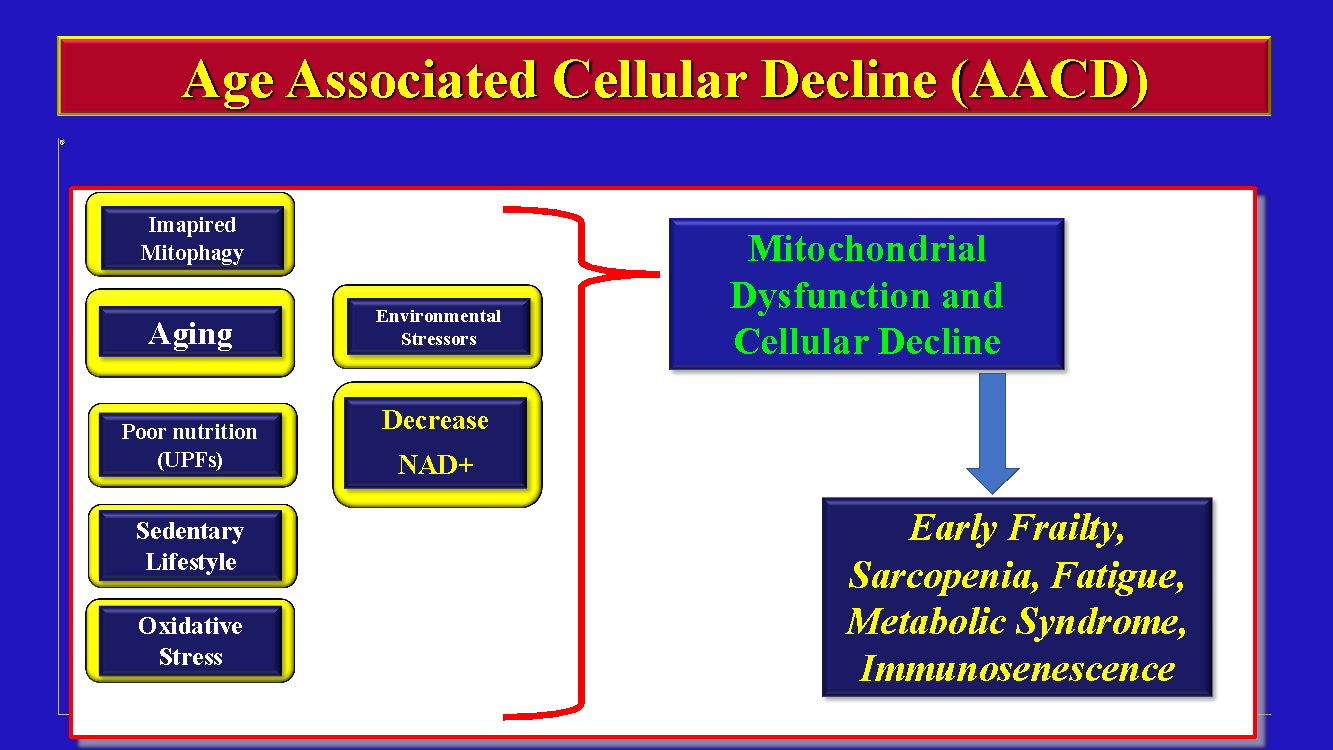
The current theory of the etiology of INRs point towards multiple mechanisms working together synergistically as seen in Figure 1.

The detailed explanation of each one of the above mechanisms of INRs are beyond the scope of this review and can be found elsewhere.1–4
The objective of this review is to describe a hypothesis of the etiology of immunological failure of PLWH on ART based on the possible role of the overactivation of the CD38 receptor with subsequent NAD+ depletion.
The CD38 Receptor and HIV Progression
The CD38 receptor is a well-recognized cellular receptor (type II transmembrane glycoprotein) detected on many cell types (but mainly on immune cells) with an extracellular domain with enzymatic catalytic activity which overactivation has been recognized as an excellent predictor of HIV progression (along with HLA-DR).5 Of note, CD4 T-cells with CD38 activation enhances nearby CD4 T cell activation generating new HIV targets for de-novo infection in a vicious cycle loop. CD38 has many functions including ADP-ribosyl cyclase, dehydrogenase, maturation of B cells in the germinal centers, activation and proliferation of T cells and NK cells, production of IL-1, IL-6, Il-10, IFN-gamma, TNF-alpha, and GM-CSF.5
There are studies showing that enhanced CD4 T cell activation can lead to further T-cell activation, high T-cell turnover, T-cell immune exhaustion, and finally, enhanced apoptosis.6 Of note, the CD4 T-cell loss induced by immune activation exceeds the one expected due to direct viral cellular cytotoxic effects.5
“….The decrease of the overall immune-activation (CD38+ / HLA-DR) after the initiation of ART is a better predictor of the CD4+T-cell recovery than the decrease on the HIV viral load5….”
ART significantly decreases the immune activation status with reduction of the CD38(+)/CD8(+) T-cell lymphocytes ratio5 which happens in all the cell subsets (CD4+ memory, CD4+ naïve, CD8+ memory, and CD8+ naïve cells).
“… there is a negative correlation between CD38(+)/CD8(+) and CD4(+)/CD8(+) ratio after starting ART5…”
Because of the above it is really important to start ART EARLY in the course of the infection not only to decrease the reservoir size but to drop the level of immune activation almost to the level of uninfected individuals.7 It is interesting to note that CD38(+)/CD8(+) T-cells from patients on ART have some protective mechanisms against apoptosis (IL-7 mediated) compared with untreated patients which allow them to persist more, and, may be, to contribute more to chronic inflammation. Interestingly, elite controllers (ECs) produce a specific type of CD38(-)/HLA-DR(+)/CD8(+) T cells which may promote low levels of immune activation, hence, generating less de novo HIV targets with less progression of disease and systemic inflammation8 which is, in fact, protective. In fact, the creation of specific CD38(-) / HLA-DR(+) / CD8(+) T-Cells may be the Goal of Future HIV vaccines or therapeutics.
“…. Based on the above seems clear that the decrease of the CD38(+)/CD8(+) T-cell ratio should be an important therapeutic target for intervention besides ART5….”
Even though we know that CD38(+) is a good predictor of HIV progression little is known about the specific role of this transmembrane protein on HIV pathogenesis. The specific mechanism of how CD38+ may lead to CD4+ T-cell depletion is not fully understood.
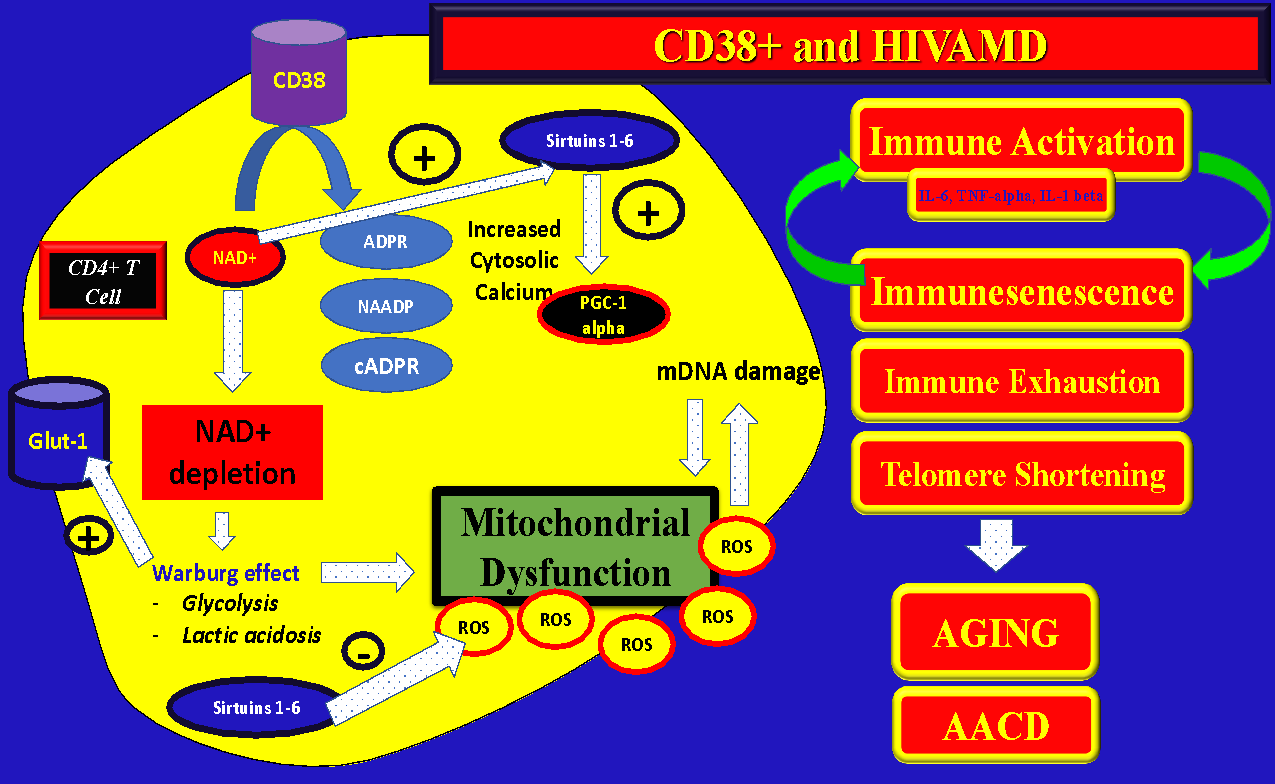
On Figure 2 it can be appreciated the multiple enzymatic functions of CD38+ specially the ones related to NAD+ metabolism, Sirtuin activation, immune-metabolic functions, and mitochondrial function regulation. CD38(+) overactivation may be in part responsible for the accelerated aging process that is seen in well controlled HIV patients on ART along with progressive mitochondrial dysfunction and frailty. The specific complex functions of Sirtuins are beyond the scope of this article and can be found elsewhere.9–18

Please note that the enzymatic activity of CD38(+) consume large quantities of NAD+ and generates “second messengers” like ADPR, NAADP, and cADPR. NAD(+) is also a co-factor of Sirtuins which are extremely important molecules for cell homeostasis, mitochondrial respiration (with decrease in the generation of mitochondrial ROS), and DNA repair.19
“…NAD(+) is extremely important to promote healthy OXPHOS (Oxidative Phosphorylation) in the mitochondria for effective ATP generation. With NAD(+) depletion, the cellular metabolism (even from immune cells) is switched to predominantly AEROBIC cytosolic glycolysis, which is highly pro-inflammatory (Warburg-effect) and much less effective for cellular energy production19,23…”
Interestingly, it was found on memory cells of PLWH an increase of the activity of the NAMPT (Nicotinamide Phosphoribosyl Transferase) which is the rate limiting enzyme for NAD(+) generation (Salvage pathway) which could be a compensatory response to the depletion of intracellular stores due to overactivation of CD38(+).22 Some of the 2nd messengers from NAD(+) metabolism can also affect the mitochondrial function due to increase cytosolic calcium affecting the integrity of the mitochondria.
In summary, overactivation of CD38(+) on CD4+ T-cells of PLWH may cause:
-
Decreased intracellular NAD+ with impairment of ATP production
-
Increase of pro-inflammatory cytokines with subsequent systemic inflammation and secondary mitochondrial damage due to oxidative stress (ROS accumulation) through structural damage (damage of mDNA, lipids, and other mitochondrial structures).
-
Persistent immune-activation can promote “immune exhaustion”. Exhausted immune cells can switch from OXPHOS to Aerobic-Glycolysis (metabolic reprogramming) which can be detrimental for mitochondrial function due to increased glycolytic / pro-inflammatory pathways (“metabolic switch or Warburg Effect”).26
-
Secondary catalytic products of NAD+ through CD38(+) enzymatic activity (ADPR, cADPR, NAADP) can open calcium channels and decrease mitochondrial integrity with apoptosis.
-
Immune exhaustion through the PD-1/PD-L1 signaling pathway can potentially also impair mitochondrial function and cell viability.
“…of note, pharmacologic inhibition of CD38 dramatically increases the levels of NAD+ in normal and obese mice24…”
Chronically activated CD4(+) T-cells show a pattern of increased aerobic glycolysis, increase of the concentration of Glut-1 transporter, and increase lactate22 (Figure 2).
Overactivation of Indoleamine 2,3 dioxygenase 1 (IDO-1) through NAD+ depletion during HIV infection
The Kynurenine pathway has been highly conserved between species.24 IDO-1 plays a central role in HIV pathogenesis through its immunoregulatory effects which can be reviewed elsewhere.27–35 For more details about the specific role of IDO-1 on HIV pathogenesis please see our recent review.36
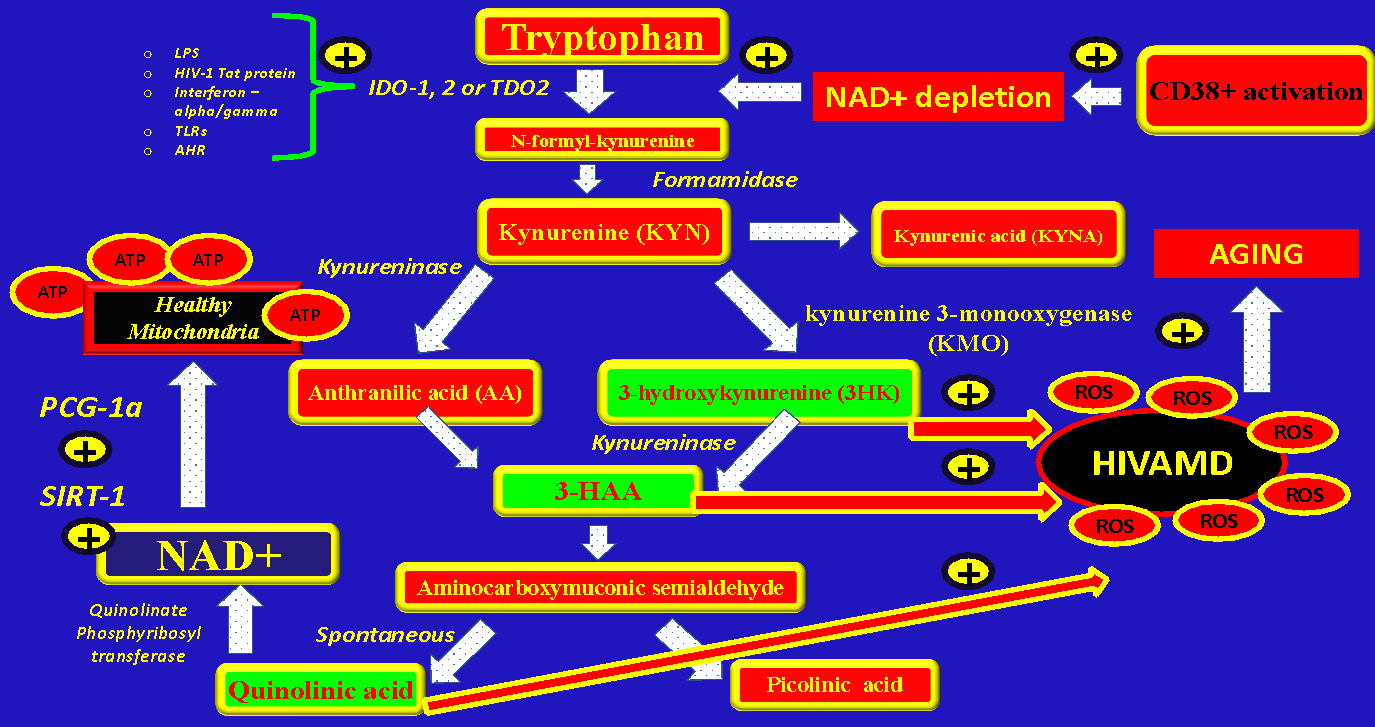
IDO-1 can be stimulated by LPS, HIV-1 Tat protein,37 IFN-alpha/gamma, Aryl Hydrocarbon Receptor (AHR) activation, Toll-like receptors, TNF-alpha, NAD+ depletion, and loss of Th17 cells. IDO-1 can be increased in many immune cells like macrophages, glial cells, antigen-presenting cells, lymphocytes, and natural killer cells (NKc).24 A study showed that there is a correlation between IDO-1 activity and HIV DNA in blood, Immune-activation, and T cell exhaustion.29 IDO-1 catalyzes the conversion of Tryptophan to N-formyl-D-kynurenine and it’s metabolites and increases the Kynurenine/Tryptophan ratio. Also IDO-1 promotes the conversion of naive T cells to Tregs,29 hence, it can modulate the immune response.
In the kynurenine metabolic pathway starting from Tryptophan (TRP) one of the last steps is the generation of NAD+ from Quinolinic acid due to the enzyme Quinolinate phosphoribosyl transferase (de-novo NAD+ synthesis pathway). Any disbalance on the amount of available tryptophan or its intermediate metabolites could negatively impact the generation of NAD+ from quinolonic acid (see Figure 3).

An increase in the activity of IDO-1 seen during HIV infection could deplete the amount of tryptophan available for the generation of de-novo NAD+ in order to generate other intermediates metabolites (some of them causing mitochondrial damage like 3HK, 3-HAA, and QA), hence, causing a decrease in the total pool of NAD+ and CD4 T-Cell depletion
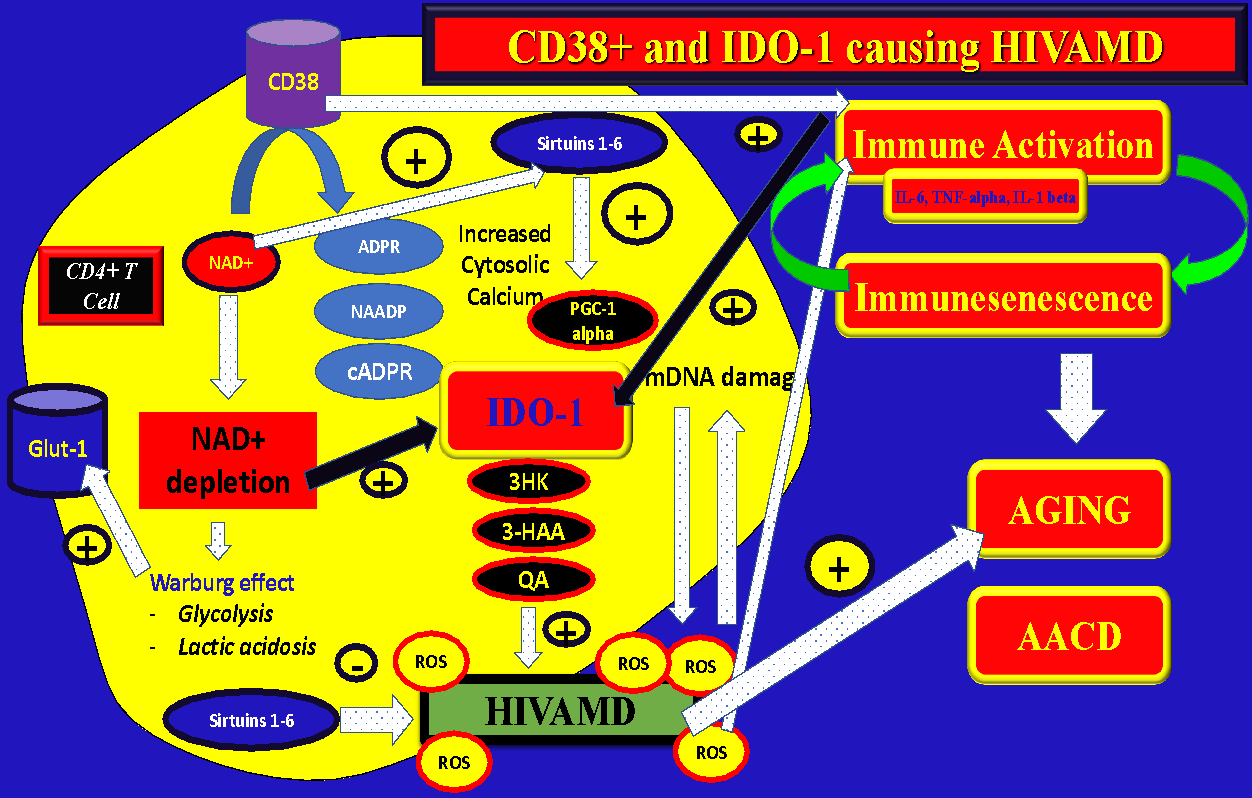
Two hypothesis are feasible to explain immunological failure: CD38 activation –> NAD+ depletion –> IDO-1 activation –> HIVAMD –> Apoptosis (CD4 decline):
-
Hypothesis #1 = Overactivation of CD38 on PLWH produce a depletion of NAD+, which, in turn, accelerate the IDO-1 metabolic pathway in order to generate de-novo NAD+. The increased flow of reactions through the Kynurenine pathway could overproduce the intermediate metabolites (instead of NAD+) 3HK, 3-HAA, or QA causing HIVAMD, NAD+ depletion, and cell death…." (Figure 4)
-
Hypothesis #2 = Overactivation of CD38 during HIV infection generates immune activation with increased secretion of pro-inflammatory cytokines (especially TNF-alpha) with subsequent activation of IDO-1 not mediated by NAD+ overconsumption (inflammatory pathway). (Figure 4)

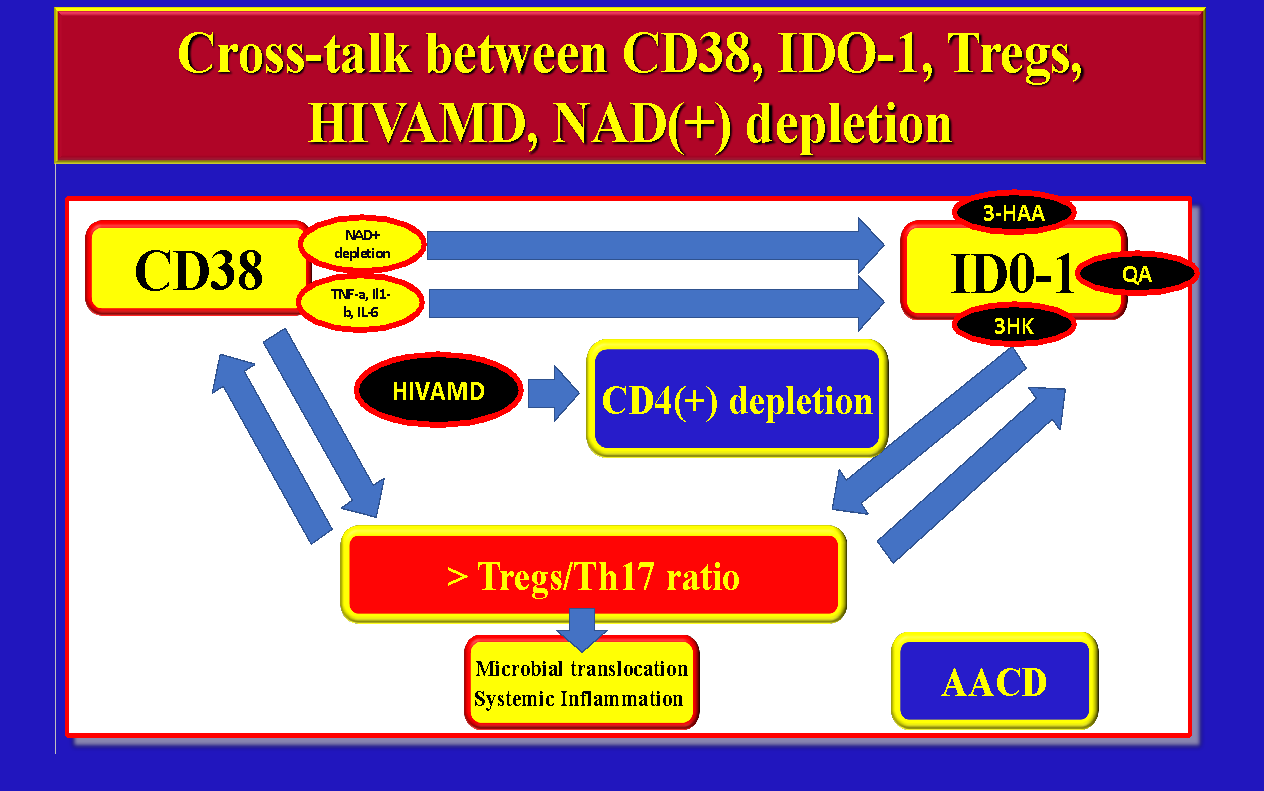
Based on the above we can conclude that CD38(+) overactivation, NAD(+) depletion, IDO-1 activation, Immune-activation, HIVAMD, microbial translocation, and CD4(+) T-Cell apoptosis may be all related and interconnected (Figure 5).

NAD(+) Depletion and Aging
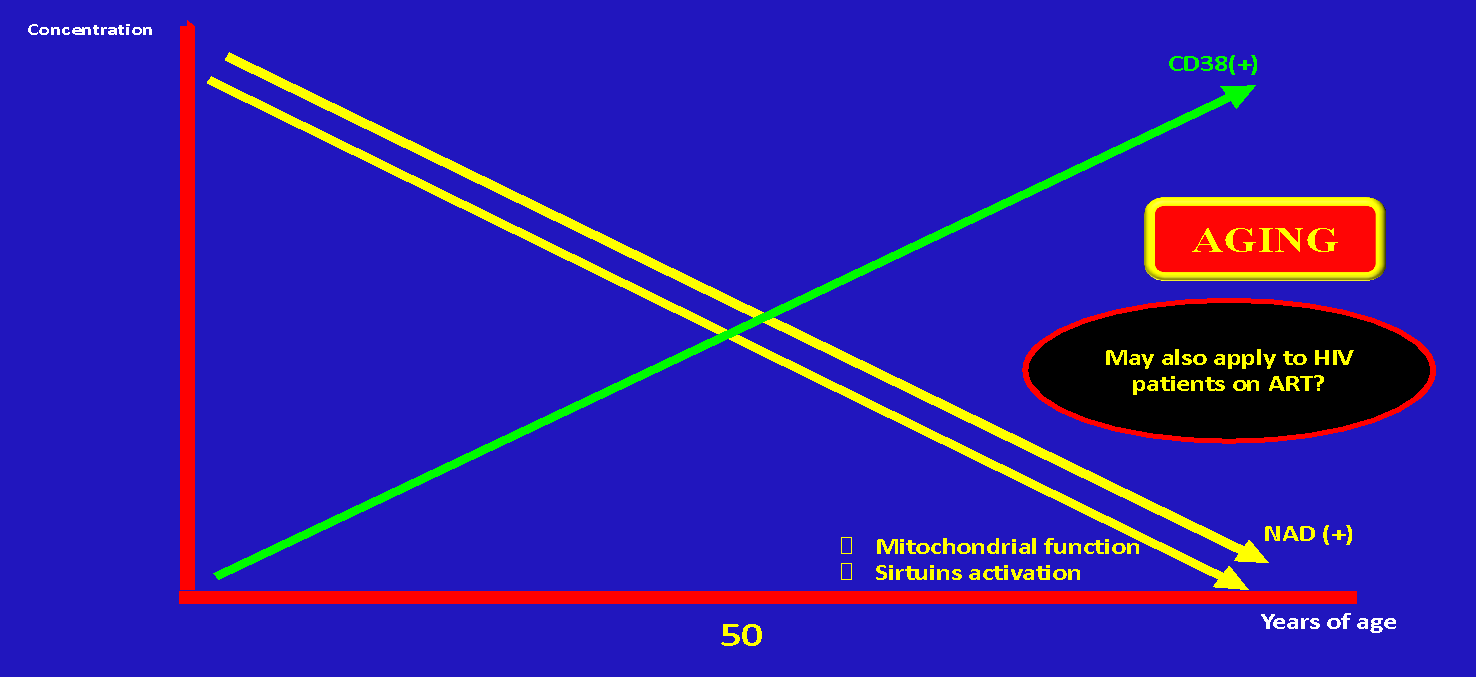
There is a clear decline of NAD+ with age which may have profound metabolic consequences promoting accelerated aging, AACD, and decline in cellular energy production (Figure 6).

Of note, NAD+ declines up to 50% between 40 and 60 years of age23,25,38–40 (Figure 7). At the same time with aging there is a progressive increase of CD38.

Given the fact that well-controlled PLWH have accelerated aging the question is if CD38(+) overactivation and NAD(+) depletion may explain not only the ultimate cause of immunological failure in a subtype of patients but the accelerated aging on well controlled individuals on ART as well.
Possible interventions: A Pragmatic Approach to Reverse Immunological Failure with a Geroscience-guided Approach
People living with HIV (PLWH) are experiencing a disproportionately high incidence of age-associated diseases, often seen in more senior demographics. This suggests that strategies from geroscience applied in certain clinical trials may also benefit this demographic. Central to the pathogenesis of immunological failure (IF) could be CD38+, IDO-1 activity, HIVAMD, and NAD+ depletion. To counteract, and possibly reverse, the IF in some PLWH, it’s imperative to investigate novel immune-metabolic and nutritional avenues.
It is apparent that antiretroviral therapy (ART) only mitigates some metabolic and immunological anomalies in PLWH, primarily curbing extensive HIV replication in plasma and averting the progression to AIDS. We propose a hypothesis linking HIVAMD, CD38, IDO-1, chronic inflammation, and immune activation to the phenomenon of IF, highlighting NAD+ depletion and OXPHOS pathway deficiencies as key contributors. There is an urgent need for future randomized controlled trials to test these hypotheses and to seek ways to alleviate or enhance outcomes related to IF and accelerated aging in PLWH. Ground-level, practical interventions involving diet, lifestyle changes, and medications (including dietary supplements) should be initially tested in community settings, such as local hospitals and non-profit organizations. These smaller, pragmatic, proof-of-concept studies could lay the groundwork for larger, more extensive trials in academic institutions.
Potential practical studies
-
NAD+ supplementation: Utilizing precursors like NMN or Nicotinic acid.
-
CD38 inhibition through supplements: Exploring the potential of Apigenin, Quercetin, and Luteolin.
-
Pharmacological CD38 inhibition: Investigating in closely monitored clinical trials.
-
Sirtuin activators: Utilizing compounds like Resveratrol.
-
Mitochondrial function enhancers: Investigating supplements like Urolithin-A or Methylene blue (only in controlled clinical trials).
-
Caloric restriction through intermittent fasting: Exploring its potential for improving mitochondrial function and NAD+ generation.
-
Pharmacological interventions with senolytic and senomorphic agents: Considering options like Dasatinib plus Quercetin, Fisetin, Bcl-2 inhibitors, Metformin, and mTOR inhibitors (Rapalogs), most of them only in closely monitored clinical trials.
-
Manipulation of the kynurenine pathway: Specifically targeting IDO-1 enzyme activity and its intermediate metabolites and enzymes.
-
Microbiome modulation: Utilizing prebiotics, probiotics, and fecal transplants.
Strengths and Weaknesses of the Manuscript
Strengths
-
Novel hypothesis: The manuscript proposes a novel and potentially groundbreaking hypothesis linking CD38 overactivation, NAD+ depletion, and mitochondrial dysfunction to immunological failure in PLWH.
-
Integrative approach: It integrates various aspects of HIV pathogenesis, including immunology, metabolism, and aging, offering a comprehensive perspective.
-
Translational potential: The manuscript highlights potential therapeutic targets and interventions based on the proposed hypothesis, paving the way for future research and clinical trials.
-
Geroscience perspective: Incorporating principles of geroscience allows for a broader understanding of accelerated aging in PLWH and its connection to IF.
Weaknesses
-
Hypothetical nature: The proposed mechanism is primarily hypothetical and requires further experimental validation.
-
Limited direct evidence: While supported by existing literature, direct evidence linking CD38 overactivation to NAD+ depletion and mitochondrial dysfunction specifically in the context of HIV immunological failure is limited.
-
Complexity: The interplay of various factors involved in the proposed mechanism is complex and requires further elucidation.
-
Specificity: The proposed mechanism may not be universally applicable to all INRs and may only be relevant to a subset of patients.
Conclusion
The manuscript presents a compelling hypothesis, offering a new perspective on the elusive etiology of immunological failure in virologically suppressed PLWH. However, rigorous experimental validation is necessary to confirm the proposed mechanism and assess the efficacy of potential interventions. Exploring this hypothesis could potentially revolutionize our understanding of immunological failure and pave the way for novel therapeutic strategies aimed at restoring immune function and improving long-term health outcomes in PLWH.