Introduction
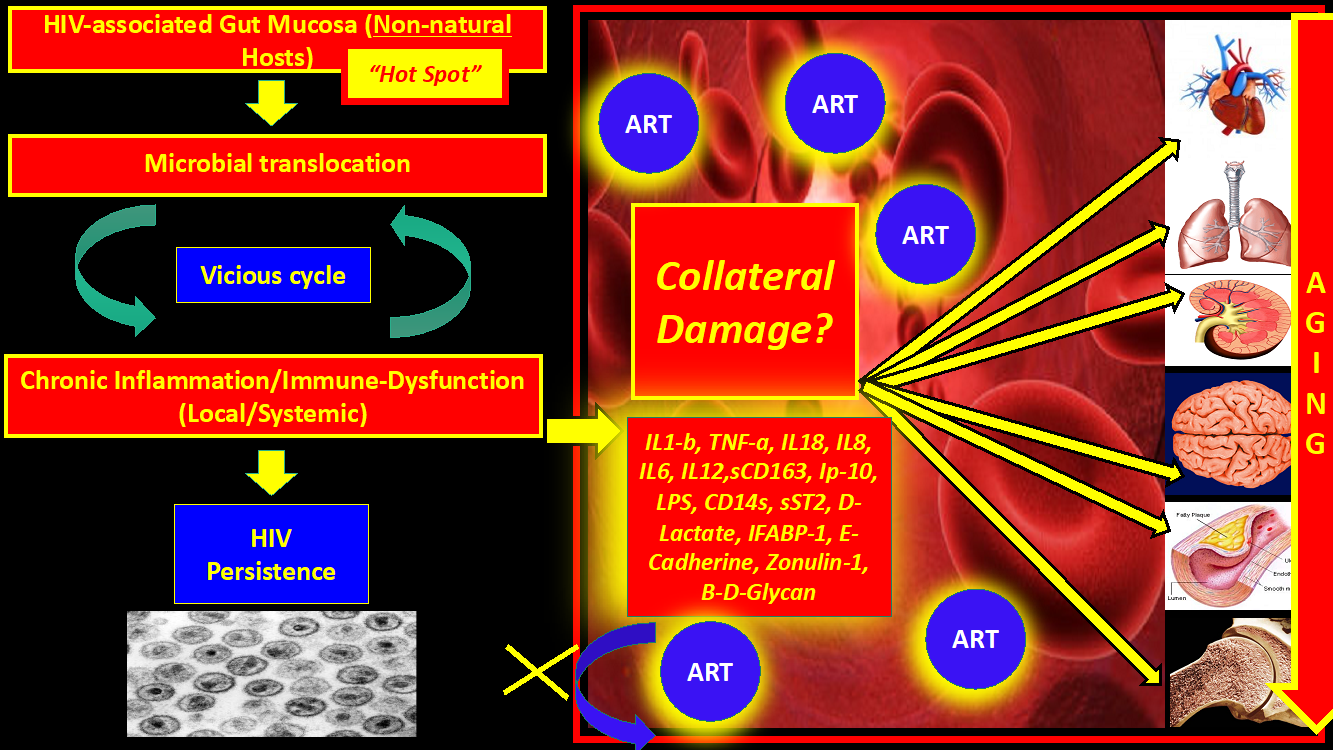
Owing to the introduction of successful antiretroviral therapy (ART) since the 1990s, we have turned Human immunodeficiency virus (HIV) into a chronic infection. Despite effective regulation of viral replication in plasma and a significant decrease in the overall mortality, there is increasing evidence that even HIV-infected patients who are considered “optimally treated” have a high prevalence of non-AIDS- defining illnesses (cardiovascular, respiratory, neurologic, metabolic, renal, and liver disease) along with different types of solid and hematologic malignancies.1–12 There are many hypotheses about the genesis. Most of the resulting chronic organ damage appears to be linked to a generalized state of chronic inflammation and persistent immune activation. These observations have led to the concept of "HIV and aging "or “Inflammaging”, where virologically-suppressed HIV patients on ART experience progressive and disseminated organ damage in comparison to matched HIV negative individuals , with a faster progression to aging-related organ dysfunction.2,4,5,9 Interestingly, “Elite-controllers”, who possess virologic and immunological control without ART, still suffer from chronic inflammation and aging, which implies that Inflammaging may be a direct consequence of the inflammation itself and, at least in part, somewhat independent from virologic factors. From an evolutionary perspective, “aging” could also be seen as an unintended consequence or collateral damage of HIV in order to reach the primary goal of Lentiviruses: DNA integration and persistence (Figure 1). Of note, Lentiviruses may actually need to avoid the aging process in the host in order to persist longer since aging decreases survival. Because of the problematic issues detailed above, multiple clinical trials, proof-of-concept studies, and research theoretical models have been published with the ultimate goal of decreasing inflammation and aging in HIV patients on ART, whose results have been reviewed extensively elsewhere13–41 and have been disappointing.

The objective of this review is to focus the analysis of the pathogenesis of HIV on the gut mucosa as epicenter and origin of the persistent local and systemic inflammation, immune activation, microbial translocation, and aging. We will also discuss some similarities between HIV and Simian immunodeficiency virus (SIV) in non-natural hosts (NNHs), since this has traditionally been the experimental model used to study HIV pathogenesis.
Introduction to the HIV- Associated - Gut Mucosal Dysfunction: The Goals of Reversal of the "Dysbiosis "and Promotion of the Epithelial Gut Integrity
Microbial translocation
There is overwhelming evidence that persistent microbial translocation in a dysfunctional and structurally abnormal GI mucosal barrier plays a primordial role in HIV pathogenesis.42–50 Epithelial damage and microbial translocation are of paramount importance as the initial triggers of chronic inflammation and have been reviewed in detail previously.51–56 Some of the mucosal changes occur early in the course of infection and are only partially reversed by ART. It is known that HIV initially targets specifically CCR5+CD4+ cells in the GALT.57 Of note, intestinal lymphocytes represent over 60% of the total pool.58 There is a massive loss of CD4+ cells in the first 7 days post-infection, with 90% being wiped off at 2 or 3 weeks (mainly activated memory cells).58 These early mucosal changes will be so important for the pathogenic HIV infection model moving forward that we think we should consider chronic HIV infection, at least in part, as a “Chronic Inflammatory Intestinal Disease”. In fact, there are multiple similarities between Inflammatory Bowel Diseases (IBD) and HIV with regards to microbial translocation and chronic systemic inflammation.51 However, during IBD there is no depletion of mucosal CD4+ cells; on the contrary, there is an increase influx of those cells, including Th17,58 which means that gut inflammation and microbial translocation without viral replication are not sufficient to cause CD4+ mucosal depletion and immunodeficiency. In addition to viral factors, in HIV there is a correlation between microbial translocation, CD8+ cells expressing CD38 (immune activation), and failure of CD4+ immune reconstitution after ART.57 We proposed in our previous review59 that through thousands of years of evolution, HIV has learned how to artificially create these “gaps” in the tight junctions between the epithelial cells with the ultimate goal of feeding off the local inflammation and promoting local viral persistence, one of the main objectives of the Lentivirus family.59,60
…..May have HIV learned over thousands of years of evolution how to artificially create gaps in the tight junctions early during the infection in order to feed off from inflammation / immune-activation with the goal of creating new targets for “de-novo” infection cycles?…… Is the “aging” process the downside? A miscalculation?…..
In fact, a recent experiment with Caco cells showed that the HIV-1 itself, through the gp140 protein, can cause these changes early in the infection with a decrease in Claudins (CLDN), Occludins (OCLN), and Zonulin (ZO-1), which could be reversed with administration of IL17A-F. In another study, plasma levels of IFABP and sST-2 were elevated in a cohort of 48 recently infected HIV-positive patients confirming the early onset of enterocyte damage with the release of their structural proteins.53 There is a decrease in the amount of the de novo synthesis of ZO-1, Claudins 1-2, and E-cadherin early after the initial infection. The initial epithelial damage is similar to the damage caused experimentally by Acrolein, which in a study caused a decrease in zonula occludens-1, Occludin, and Claudin, in addition to inducing endoplasmic reticulum stress-mediated death of epithelial cells,61 favoring microbial translocation and immune activation. To support the above statement, a very revealing recent study showed through proteomic analysis that as soon as 3 days after SIV infection there was a decrease in proteins related to epithelial integrity and that 14 days after the infection there was an increase in proteins related to the innate immune system, which is evidence that epithelial damage occurs before the innate immune response.54 In some experimental studies, as soon as 14 days after primoinfection, there is an almost complete depletion of CCR5+CD4+ T memory cells, which persists if it remains untreated.58 In fact, it is possible to measure in plasma an increase of D-lactate, IFABP-1 (Intestinal Fatty Acid Binding Protein), REG3-alpha62 (also increased in IBD), and sST2 in well-controlled HIV patients on ART. Of note, IFABP-1 has been linked with increased mortality and CD4+ cell decline in HIV.63 Interestingly, a recent study found that circulating Beta-d-Glucan (BDG) has a better correlation with cardiovascular disease (measured with cardiac computed tomography) than LPS.64
"….An increase in plasma REG3-alpha (a marker of gut damage) is increased in PLWH (whether on treatment or not) including elite controllers compared with HIV negative controls. REG-3-alpha levels correlate inversely with CD4 cell count and positively with HIV viral load.65
How natural Hosts of SIV Resolved the Problem
On the other side, natural hosts of SIV (Sooty Mangabeys, African green monkeys, madrills, drills, and sun-tailed monkeys) don’t have chronic inflammation, microbial translocation, immune activation, immune exhaustion, AIDS, or accelerated aging.66 The CD4+ cells of natural hosts during SIV infection have a slight initial decrease after the initial acute infection, and soon thereafter pre-infection levels are reached with a downregulation of CCR5, which decreases infections de novo.58 Natural hosts adapted and learned how to control cellular depletion and inflammation, one known mechanism being the decrease in the number of HIV-target cells like CCR5-CD4 T lymphocytes in the GALT.58
Intriguingly, if you give high-fat diets to natural hosts of SIV (African green monkeys) they develop progressive disease with increase of microbial translocation, immune-activation, viral replication and expansion of the reservoirs with progressive disease.67
SIV in natural hosts will significantly decrease the inflammation and immune activation after the transition from acute to chronic SIV infection, in spite of persistent and robust viral replication, which highlights the fact that viral factors may not be as important as immunomodulatory factors for CD4+ cell depletion and disease progression.58 This has been achieved due to enhanced gut mucosal repair mechanisms associated with a decrease in microbial translocation. It was also seen that in natural hosts, a mutated gene of ICAM2 and TLR-4 impairs the response to LPS stimulation with a subsequent decrease in the production of TNF and IL6 and local and systemic inflammation by the local innate immune system.68 Also, sooty mangabeys macrophages were found to be more resistant to infection by SIV than their rhesius macaques counterparts through an increase in the expression of Tetherin and TRIM22.69 Another novel mechanism of protection against SIV in natural hosts is the downregulation of interferon responses, which is associated with pathogenic SIV and HIV and causes immune activation, immune exhaustion, and apoptosis.58 More evidence to support the fact that viral factors alone don’t explain the genesis of immune activation and inflammation is that even though elite controllers have immunological and virologic control without ART, they still have some degree of epithelial damage since their IFABP-1 levels in plasma are lower compared with HIV progressors but still higher than HIV uninfected controls.63
In summary, this artificially-created “Hot Spot” (as we called the GUT-mucosa and GALT – HIV/SIV-associated changes in non-natural hosts) (Figures 1, 2) soon after the infection will be fed continuously with an enormous amount of antigens, chemicals, bacteria, fungi, viruses, andenvironmental substances, which in turn, promote persistent activation of the innate immune system in the context of an already abnormal microbiome (see below “Dysbiosis”). Bacterial and fungal (and, may be, viral) antigens continuously cross-over through the gaps without immunological control or “check-points” even after being on ART. Tight junctions damage is the initial tissue insult or “spark” that will trigger inflammation and immune activation.
The central role of Th17 cells and MAIT cells
A rapid decline in Th17 cells (CCR6 positive) after acute pathogenic HIV infection with an increase in the ratio Tregs/Th17 is believed to be the culprit of the persistent lack of epithelial homeostasis and lack of repair of the tight junctions after the initial damage51,53,55,60,66,70,71 and is linked to inflammation and immune activation.58 Of note, the Tregs are also important reservoirs for HIV.72
“….The Th17/Treg ratio remain stable during chronic SIV infections in natural hosts while it is decreased in pathogenic HIV/SIV infections and predicts disease progression58…”
Tregs increase the “tolerance” to microbial products leakage since their main function is to suppress “regulate” the immune response, usually against self-antigens.53 The mechanism used by HIV-infected Tregs to regulate immune activation may be mediated through generation of local adenosine (Purinergic pathway).72 Unfortunately, Th17 cells are only partially restored with ART in blood and not restored at all in GALT (Gut Associated Lymphoid Tissue), which seems an irreversible phenomenon.55 As a result, HIV can decrease the production of IL-17 and IL-22, which are highly involved in the repair of tight junctions and barrier integrity.58 IL-21-producing CD4+ T lymphocytes are also depleted, which can alter immune pathways that would favor Th17 differentiation.58 Another group of local cells that assist in the mucosal repair mechanisms are the MAIT cells. One study showed that activation-induced pyroptosis is responsible for the loss of MAIT cells in the GI mucosa of PLWH (even on ART) compared to HIV negative controls.73 MAIT cells are responsible of the integrity of the epithelial barrier against external challenges like microbial or fungal translocation. The above study also showed that the absolute number of MAIT cells correlated inversely with plasma sCD14+ (marker of monocyte activation due to microbial translocation) and I-FABP levels in HIV treatment naïve patients.
The Vicious Cycle that Never Ends
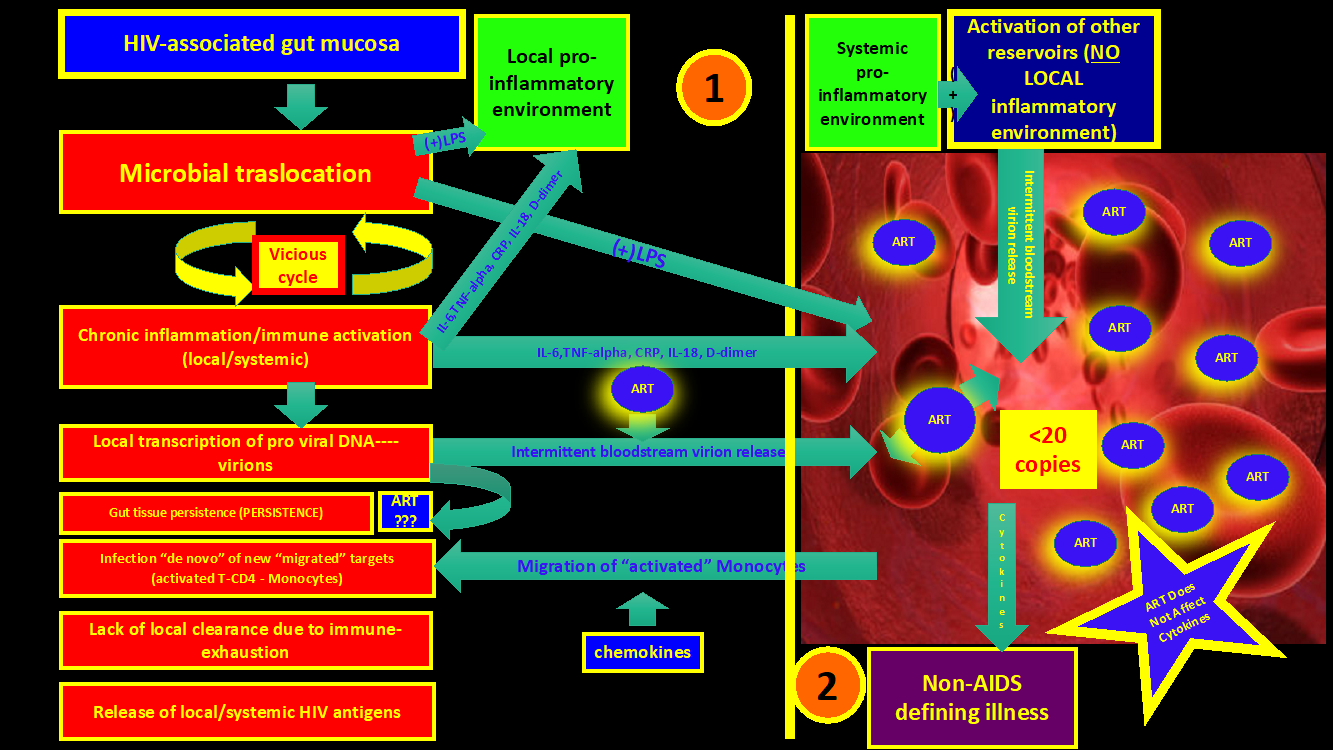
“….The above pathogenic mechanisms represents a “vicious cycle” on which the persistent microbial translocation through a “leaky epithelium” of an already Dysbiotic microbiome continuously stimulate an already exhausted and senescent innate and adaptive immune system (Figure 2 and 3)….”
The leakage will promote chronic inflammation and immune activation with local HIV persistence since immune cells are constantly activated, generating new HIV targets. It is likely that ART, even though it may not optimally penetrate the GUT mucosa and GALT, is effective enough to control the gross active HIV replication in plasma. Of note, the inflammatory signals (Interleukins, chemokines, etc.) and microbial products already have reached the bloodstream at that point and will deliver the initial inflammatory trigger “spark” in distant reservoirs (the CNS, cardiovascular, renal, bone, etc.), accelerating the aging process and organ damage.
"…..To close the tap (leakage) is the only way to end the vicious cycle of microbial translocation, inflammation, and immune activation…..?


“Warburg-like” Phenotype in Immune Cells: The Immune-Metabolic Switch.
Based on published evidence, it is clear that the local gut inflammatory environment in HIV patients promotes glycolysis as the preferred source of energy and the exocytosis of its products on immune cells.51–53,55,56,71,74,75 This phenomenon is called “metabolic reprogramming” and implies that immune cells go through a “broken Krebs cycle” with accumulation of Citrate and Succinate, both of which are very pro-inflammatory. The final result is the activation of the NLRP3 inflammasome and its consequences.76 Briefly summarized, the genesis of the increased glycolytic pathways in HIV may be related to multiple factors: an abnormal microbiome (“pro-glycolytic” dysbiosis), strictly nutritional factors (diet rich in carbohydrate and saturated fats), preferred (selected through evolution) glycolytic pathways during inflammatory states, or a combination of the above. The process starts when through molecular signaling there is a constant recruitment of metabolically active monocytes (who express more Integrin B7) from the bloodstream to the “hot spot” (Gut-mucosa). The chemokine monocyte chemoattractant protein 1 (MCP1) or CCL2 is mainly responsible for the recruitment. Those monocytes will overexpress Gut-1 transporters which favors the accelerated internalization and overutilization of glucose. The immune-metabolic phenotype change also affects the lymphocyte subclass CD4+ PD-1 / CTLA- 4 which through mTOR pathways will promote further local HIV persistence. The increase availability of glucose locally (intracellularly and extracellular through exocytosis in vesicles) will also favor the phenotype switch of bystander immune cells (especially APCs and macrophages) to the M1 phenotype or “Warburg-like” (Glycolytic or Pro-inflammatory).51 The hyper-glycolytic environment will promote HIV persistence and a significant increased secretion of TNF-alpha, IL-6, and CXCL2.51 Actually, the increased translocation of LPS and its uptake by those cells (mainly through TLR4 pathways) will promote the switch from Oxidative phosphorylation (OXPHOS) to glycolysis which shows proof of a metabolic-driven immune-phenotype change. This topic has been extensively explored in a recent review77 showing that the proportion of CD4+Glut1+ cells correlated with an increase on immune activation defined by expression of HLA-DR and CD38 and inversely correlated with the absolute CD4+ cell counts. There is a clear association between T cell glycolysis activity and increased susceptibility to HIV infection which was demonstrated in multiple studies from different groups and was proven by the impaired viral infection due to inhibition of glycolysis in some invitro studies.
“…..Intriguingly, immune-metabolic changes were also documented on “HIV elite controllers” but with fewer metabolomes involved in glycolysis and the Krebs cycle and favoring lipid metabolism on specific HIV-1 CD8+ T cells in order to become more active and polyfunctional78…..”
Of note, on the healthy gut mucosa of HIV negative individuals the predominance of Firmicutes (with decrease of Proteobacteria) increase the local availability of Butyrate, which, in turn, due to its high volume of distribution, reaches immune cells and promotes lipid B-oxidation and OXPHOS metabolic switch with a subsequent increase of the release of anti-inflammatory and antimicrobial molecules (Oxidative-M2 Phenotype). HIV patients instead present a local increase of Proteobacteria, Prevotella, and fungus (see below under “Dysbiosis”). Their products (LPS, B-d-Glucans) promote glycolysis, secretion of pro-inflammatory cytokines, and further recruitment of Warburg-like monocytes from the bloodstream in a persistent-endless vicious cycle.51 The HIV itself through its Vpr protein can stimulate this immune-metabolic change through the HIF-1 alpha (stress response to hypoxia) and PPAR-gamma pathway.51 Some theories also point towards the potential for the HIV itself to stimulate HIF-1 alpha pathways in lymphocytes with secretion of glycolytic products promoting cell exhaustion to local bystander cells.51 In summary, glucose availability and glycolytic pathways are important players on the genesis and maintenance of a local and systemic inflammatory environment and HIV persistence.
Indoleamine 2,3-dioxygenase 1 (IDO-1)
It has been known for a number of years now that the Kynurenine (KYN)-to-Tryptophan (TRP) ratio increases with aging, some neurodegenerative diseases, and on chronic HIV infection.79 IDO-1 is believed to play a central role in HIV pathogenesis through its immunoregulatory effects which has been extensively reviewed elsewhere.51–53,55,56,70,71,74,75 This enzyme transforms TRP to KYN and increase the KYN/TRP /Tryptophan ratio. Also IDO-1 promotes the conversion of naive T cells to Tregs74 which is detrimental in HIV pathogenesis (see above). There is an increase in IDO-1 activity in some difficult-to-reach sites like testicles, eyes and brain, which shows that it may also be involved in HIV persistence also in “Sanctuary sites”. There is a direct relationship between increase IDO-1 activity and increase on IL-1b, IL-6, IL-18, IP-10, TNF-a, sCD40L, LPS, and sCD14.76 An extensive review of the literature showed this enzyme to be the focus of multiple clinical trials as a possible therapeutic target, even in cancer research. LPS, HIV-1 Tat protein,80 IFN-alpha/gamma, Aryl Hydrocarbon Receptor (AHR) activation, Toll-like receptors, and loss of Th17 cells are between the promotors of the activity of this enzyme. Increase IDO-1 activity has been linked to increase pro-viral DNA, increased viral reservoir, poor CD4+ recovery, HIV progression, AIDS and non-AIDS events, increased mortality,52,74 increase on cardiovascular disease, reduced cognitive performance, and increase frailty.79 After 2 years of effective ART HIV patients with high N-formyl-D-kynurenine in plasma had higher CD14’s, higher percentage of Tregs, lower percentage of naïve cells, lower CD4/CD8 ratio, decreased amount of Th17 cells,58 and poor immune reconstitution compared with patients with low Kynurenines.81 A recent study also showed that there is a correlation between IDO-1 activity and HIV DNA in blood (reflecting its possible influence in the reservoir size), Immune-activation, and T cell exhaustion.74 In human monocytes derived cells (MoDC) HIV-1 Tat protein induced IDO-1 expression and activity in a NF-κB dependent-manner by recruiting the TLR4 pathway.80 In a recent very interesting study a significant decline of serotonin along with an increase on KYN and IDO-1 activity was seen on older PLWH79 which means that the IDO-1 activity in PLWH that are aging shift towards the KYN pathway instead of the serotonin pathway.
The Dysbiosis present on pathogenic SIV/HIV models with depletion of Lactobacillus and increase in Proteobacteria and Prevotella (see below under “Dysbiosis”) could also promote IDO-1 activity since it was seen that Lactobacillus supplementation decreased IDO-1 activity in some experimental models82 and that dysbiosis with loss of Lactobacillus increased IDO-1 activity and correlated with Th17 cell loss.58 In fact, the supplementation of IL-22 along with probiotics could decrease microbial translocation and inflammation with decrease of IDO-1 activity.66 In the study mentioned above they found that Firmicutes and Bacteroidetes also presented quantitative changes dependent on the age in PLWH: increasing age is negatively associated with Firmicutes and positively associated with Bacteroidetes.79 The Firmicutes/Bacteroidetes ratio, a well-known marker of gut-dysbiosis, also showed a decline related to age in the same study. This is important since a decrease of the population of Lachnospiraceae and Lactobacillaceae will cause a proportional decrease of butyrate levels (see section below SCFAs)
Summary of IDO-1 role in HIV pathogenesis based on an extensive literature review83–86:
-
LPS-stimulated-TLR4 in APCs will increase the activity of IDO-1 which transforms Tryptophan to N-formyl-D-kynurenine and increases the Kynurenine/Tryptophan ratio.
-
The increased Treg/Th17 ratio will also increase the IDO-1 activity.55
-
IDO-1 will stimulate the NF-kb factor with increase in pro-inflammatory cytokines.
-
There will be also an increment of the products of Glycolysis favoring the switch to the M1 (pro-inflammatory) phenotype on Macrophages and APCs (see above section).
-
N-formyl-D-kynurenine also inhibits TH17 cells, which through secretion of IL-17 play a primordial role in tight junction homeostasis and repair (increase the production of the tight junction proteins Claudin 1-2).
-
Kynurenine also will promote the formation of Tregs (FOXp3 – CD39 phenotype) which decrease the T-lymphocyte cytotoxic cell response and promotes HIV persistence. In the GALT (Gut Associated Lymphoid Tissue) the increase amount of Tregs promote the formation of TGF-B1 which in turn increase the transcription of Collagen mRNA (promoting GALT-fibrosis), decrease the T cell function, and increase apoptosis.
-
The resulting abnormal architecture of the GALT tissue promote even further lack of HIV clearance (and also probably co-pathogens) and decrease the local immune function in general.
-
Unfortunately, the increase in Tregs and IDO-1 activity, along with GALT fibrosis persists even while on effective ART.71
-
N-formyl-D-kynurenine may also stimulate PD-1 receptors in the surface of CD4+T cells, favoring the activation of mTOR and promoting HIV persistence since data suggest that PD-1 negatively regulates T cell responses.
Microbiome alteration (Dysbiosis)
An extensive review of the literature points towards a deep alteration of the gut microbiome in HIV patients compared with HIV negative controls, even while on effective ART.51–53,55,56,70,75,87 In PLWH there is a significant increase in the Proteobacteria, Candida (probably with increase translocation of BDG) and Prevotella with decrease in Bacteroides, Firmicutes (importantly, Roseburia), Bifidobacterium, and Lactobacillus. The increase of Proteobacteria is correlated with heightened T-cell activation in the blood and gut, lower mucosal IL-17/IL-22 secretion, and higher plasma KYN/TRP ratio (see above).55 Usually an increase in Proteobacteria will translate into gut-damage and inflammation.70 Interestingly, Bacteroides and Firmicutes play an important role on the secretion of SCFAs (Short Chain fatty Acids) like Popionate, Butyric acid, Acetate, and Valeric acid which have protective effects on the GI mucosa. Of note, Roseburia (Firmicutes), who is reduced in HIV dysbiosis play an important role on the regulation of the immune system, Treg regulation, and increase in Bacteriocin-like substances (BLIS).70 Lachnospiraceae, also decreased in HIV patients, has been linked to protection against colon cancer due to its production of Butyric acid.70 As described above, in one study there was a negative correlation between age and the proportion of Lachnospiraceae in PLWH.79
“……In one study it was shown that there is a strong correlation between the loss of butyrate-producing bacteria (with decrease butyrate levels), the decrease of serotonin, and increase of KYN levels in older PLWH even on ART79…..”
Remains to be fully elucidated which phenomena occur first: dysbiosis, decreased SCFAs, or microbial translocation. Some proteomic analysis suggests that the metabolic changes (decreased SCFAs) occur actually before than the structural epithelial damage which may imply that the microbiome change precedes the tight junction lesions.70 Other evidence (reviewed above) shows instead that the structural lesions occur as soon as 3 days after infection. More research is needed to elucidate the initial mechanisms of gut mucosal damage.
The Importance of Short Chain Fatty Acids (SCFAs)
Butyrate is particularly important for colonic health because it is the primary energy source for colonocytes. Butyrate may have implications for epithelial barrier repair and integrity after the initial HIV structural insult. SCFAs help boost the protective mucus layer in the gut and also have the ability to influence genes that control cell proliferation and cell cycles. SCFAs can increase the secretion of antimicrobial peptides and the secretion of IL-18 by the epithelial cells to help keeping the anatomical and functional integrity of the epithelial barrier.70 SCFA’s primordial role on gut homeostasis was recently revealed in a trial on rats where exposure to Ceftriaxone promoted intense Dysbiosis with significant decrease of SCFAs along with increase in the gut leakage, and inflammatory cytokines (TNF and IL-10). Also it was found that circulating levels of butyric acid are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis.88 The loss of butyrate-producing bacteria using the acetyl-CoA pathway can also be seen on ethanol-induced microbial dysbiosis, a disease that also favors microbial translocation.89 In a gut inflammatory environment like HIV it was shown that SCFAs can decrease IL-12 and TNF-alpha.70 Adding to its multiple properties it has been seen that Butyric acid and Acetoacetate can also decrease inflammation through inhibition of the NLRP3 inflammasome - mediated inflammatory response with significant reductions of interleukin (IL)-1β and IL-18 production in human monocytes.90 It has been also confirmed that SCFAs (mainly Butyric acid) inhibit the activity of histone deacetylase (HDAC) [an enzyme involved in post-translational modifications, namely the process of deacetylation and the process of histone crotonylation].91 Histone deacetylase inhibitors (HDACi) are being evaluated in a “shock-and-kill” or “Purge” therapeutic approach to reverse HIV-1 latency from CD4 (+) T cells and other reservoirs. HDACi like Butyric acid could induced HIV RNA synthesis in latently infected cells and facilitate eradication after being targeted with ART or other novel therapies. Therefore, in addition to known HDACi compounds like belinostat, givinostat, panobinostat, romidepsin, and vorinostat, Butyric acid could play an important role as well. Intriguingly, the above mechanism involved unc-51-like autophagy-activating kinase 1 (ULK1) and the inhibition of the mammalian target of rapamycin (mTOR). Mechanistically it requires the formation of autophagosomes and their maturation into autolysosomes providing evidence in support of a link between HDACi, mTOR, and autophagy potentially as a valorous tool for HIV control.92
Indoles
Another critical aspect in the pathogenesis of chronic HIV and gut-inflammation is the increased production of Indols by the overgrowth of the Proteobacterias during the HIV-induced dysbiosis. Indols are a derivative from the aminoacid tryptophan. It is well known that Proteobacterias promote a Pro-inflammatory environment (see above).93 One of the consequences of the excess of Indole groups is the overactivation of the aryl hydrocarbon receptor (AHR) which, in turn, stimulates the IDO-1 with all the consequences explained previously (see above under “IDO-1”). It was seen that the AHR signaling, via IL-22, inhibits inflammation and colitis in the gastrointestinal tract of mice, which could be beneficial for epithelial integrity in PLWH.94 Of note, AHR may also inhibit TH17 cells (at least in a model of allergic rhinitis) which could be actually detrimental for the epithelial gut barrier integrity.95 As we can see AHR activation could have beneficial and detrimental consequences for HIV pathogenesis being its most significant detrimental consequence the activation of IDO-1 with the increase conversion of TRP to KYN with its immune modulatory effects.
HIV Associated Neurocognitive Disorders (HAND)
HIV-associated neurocognitive disorders (HAND) are a heterogeneous group of diseases that remain prevalent even on virologically suppressed PLWH on ART. HAND can be divided into three categories: HIV-associated asymptomatic neurocognitive impairment (ANI), HIV-associated mild neurocognitive disorder (MND), and HIV-associated dementia (HAD).96–100 The etiology of these spectrums of diseases is multifactorial and a lot of confounders (aging, depression, drug abuse, opportunistic CNS disease and co-infection with HCV) contribute to its pathogenesis. There is evidence showing an association between microbial translocation, monocyte activation and neuronal damage during HIV infection101–103 supporting the hypothesis of distal organ damage from the GI tract. Studies have also shown that dysregulation of the TRP metabolism with decrease of serotonin and increase on KYN levels are associated with neurocognitive decline during HIV infection.79 Butyrate can increase the activity of the TRP hydroxylase which promotes the transformation of TRP to serotonin which implies that butyrate producing bacteria are extremely important for cognitive health in PLWH in addition to the colonic mucosal benefits (see above, SCFAs)
“…..We see HAND only as one more part of the spectrum of consequences of microbial translocation, chronic inflammation, and immune-activation in PLWH……”
It is well known that Brain aging predispose to Alzheimer’s, Parkinson’s, and Hungtington disease.104 Interestingly inflammation also triggers insulin resistance which accelerates Neurodegeneration.105 Activation of the pro-inflammatory factor NFkb in the hypothalamus also promotes neuronal aging. Brain aging also decrease Synaptic plasticity, important for learning and storage (memory).104 The above facts with a decline in neurogenesis due to inflammation and senescence may also responsible for HAND.104 A loss of the mitochondrial effectivity and bioenergetics with increase of ROS is found in senescent tissues, and also may play a role on Neurodegeneration. Damaged mitochondria activate apoptotic programs with cell death through the NLRP3-ASC-Pro caspase-1 pathway with “pyroptosis”.104 There is plenty of evidence that inflammation plays an important role in HAND.96–101,106 Both Inflammation and oxidative stress are both responsible for Neurotoxicity in the context of HIV-1-associated dementia (HAD).76 As a poof of the above, a recent study showed that exposure of human primary microglia to ssRNA40 (HIV single strand RNA) activates the (NLRP3) inflammasome with increased secretion of the pro-inflammatory cytokines IL-1β, IL-18, and neurotoxic cytokines TNF-α and IL-1α (118). In another recent study on which HIV-Tg rats were administered LPS it was showed that HIV-1 Tat activates microglial NLRP3 Inflammasome-Mediated Neuroinflammation.84
“…..There is evidence of a known “Gut-Brain axis” in HAND which can explain how gut immune-activation and microbial translocation can potentially create cognitive impairment through the “export” inflammatory signals….”
Conclusion
There is no doubt that the initial insult early during HIV is located in the tight junctions at the gut mucosa level. Even though ART takes care of the gross viral replication in plasma its role in the control of the vicious cycle generated at the gut mucosa level is very limited. The initial insult may represent an apparent “advantage” from an evolutionary perspective generating “new targets” for “de-novo” cycles of viral infection. A miscalculation probably did not take into account the “collateral damage” created for those mucosal gaps with progressive aging of distal organs and systemic inflammation. ART will not fix the gaps or lack of check-points at the mucosal levels, will only control the massive viral replication that otherwise would be present without treatment.
“….The optimal goal would be for PLWH to emulate what through thousands of years of evolution SIV has accomplished on natural hosts in order to reach a peaceful coexistence without collateral damage…..”
This multi-compartmental model may be helpful to define new strategies in order to decrease or de-accelerate the aging process of HIV on ART. Some of the strategies that need further reasrch are:
-
To promote basic science studies to elucidate how to promote the tight junction repair through certain cell lines like Th17+ cells or certain cytokines like IL-17 or IL-22 in a sustainable way (the administration efficacy of pre-formed cytokines is short lived).
-
To define strategies to stimulate local gut stem cells in order to repair the tight junction gaps
-
To define strategies to attenuate or decrease the exuberant immunological response of the innate immune receptors (like TLR-4-3-9) to bacterial, fungal, and viral factors
-
To define strategies to increase the ratio Th17/Tregs
-
To define strategies to increase or promote “check points” at the mucosal level.
-
To promote strategies to decrease the mTOR pathway on immune cells
-
To develop strategies to inhibit IDO-1 and its products (KYN)
-
To define strategies to increase the local production of SCFAs like Butyrate
-
To define strategies to shift the metabolism of TRP to Serotonin instead of KYN
-
To define strategies to decrease the local production of Indols
-
To define strategies to produce changes on the dysbiosis generated during HIV infection promoting a more healthy microbiome with decrease of Proteobacterias and predominance of Firmicutes in a sustained way.
-
To define strategies to avoid a pro-glycolytic and pro-inflammatory environment at the mucosa level either with nutritional strategies or through pharmacological strategies
-
To define strategies to decrease the activation of the inflammasome and NLRP3 pathway at the mucosal level
-
To define strategies to decrease the recruitment of immune cells to the GI mucosa (particularly metabolically active monocytes).
-
To define strategies to limit or decrease the export of inflammatory signals from the GI tract (like CD14+) to distant organs to promote inflammation and immune activation
-
To define strategies to interrupt the Gut-brain-axis in order to decrease the incidence of HAND on PLWH on ART.